

А. Энергия активации
Большинство органических химических реакций (за исключением реакций кислот и оснований, см. с.36) протекают очень медленно, независимо от величины ΔG. Главная причина низкой скорости реакции состоит в том, что для вступления в реакцию молекулы реагента должны обладать определенной минимальной энергией, называемой энергией активации. Наглядно это можно представить с помощью энергетической диаграммы наиболее простой реакции А — > B (1). Каждое из соединений, реагент А и продукт реакции В, обладает· определенным химическим потенциалом (Pp и Рnp соответственно). Изменение свободной энергии реакции (ΔG) соответствует разности потенциалов. Для превращения в В соединение А должно преодолеть энергетический барьер, пик которого Pa выше Рp, Разность потенциалов Ра - Pp носит название энергия активации (Eа).
В пользу того, что A, в принципе, может превратиться в B, свидетельствует то обстоятельство, что Pp является средним значением потенциала для всех молекул, вступающих в реакцию. Время от времени отдельные молекулы достигают гораздо более высокого потенциала, например за счет столкновения с другими молекулами. Если в результате столкновения энергия молекулы превысит Eа, эта молекула перейдет энергетический барьер и превратится в В. На рис. 2 и 3 приведено распределение энергии для молекулярных ансамблей, рассчитанное на простой модели. Δn/n это та часть молекул, которая обладает (или превышает) энергией E (в кДж/моль). Например, при 27°С около 10% молекул обладают энергией около 6 кДж/моль. Энергия активации химических реакций обычно существенно выше. Аналогичный график для реакции с энергией активации около 50 кДж/моль приведен на рис. 3. Статистически при 27°С такой энергией обладает только 2 из 109 молекул, при 37°С — четыре молекулы (3). Подобная зависимость позволяет объяснить найденный эмпирическим путем температурный коэфициент скорости биологических процессов Q10, который означает, что при повышении температуры на 10°С скорость реакции возрастает в 2 раза.
Б. Скорость реакции
Скорость химической реакции определяют по изменению концентрации одного из реагентов или продуктов реакции за определенный период времени. В приведенном примере в 1 л раствора за 1 с расходуется 3 ммоля реагента и, в результате образуется 3 ммоля продукта. Это соответствует скорости реакции
v = ЗмМ · с-1 = 3 · 10-3 моль · л-1 · с-1
В. Порядок реакции
Скорость реакции зависит не только от энергии активации и температуры, но и от концентрации реагентов. Если имеется лишь один субстрат А (1), то скорость ν прямо пропорциональна концентрации [A]; это реакция первого порядка. Если в реакции участвуют два реагента А и В (2), то речь идет о реакции второго порядка. В таком случае ν пропорциональна произведению концентрации реагентов. Коэфициенты k и k' — константы скорости реакции — зависят от типа реакции и условий ее проведения.
Явление катализа- Катализатор изменяет скорость реакции, сам не изменяется. Катализатором назыв.- понижает энергию активации.Ингибитор- повышает энергию активации.—это вещества, изменяющие скорость реакции за счет участия в промежуточном хим. взаимодействии с компонентами реакции, но восстанавливающие после каждого цикла промежуточного взаимодействия свой хим. состав.
Увеличение скорости катализируемый реакции связана с меньшей энергией Гиббса активации нового пути реакции.
Изменение скорости катализируемый реакции за счет снижения энергии активации ее отдельных стадий можно рассмотреть на следующем примере.Допустим, между веществами А и В возможно взаимодействие с образованием соединения АВ(∆G<0): А+В→ А…В→ АВ активированный комплекс
Но в силу высокой энергии Гиббса активации эта реакции протикает с очень малой, практический равной нулю скоросью. Пусть, найдено такое третье вещество К (катализатор), которое легко вступает во взаимодействие с А (в силу другойприроды реагирующих веществ, а следовательно , другой, меньшей, энергии Гиббса активации), образуя соединение АК: А+К→ А…К → АК активированный комплекс
Соединение АК легко взаимодействуют с веществом В (опять-таки в силу иной природы веществ и малой энергии активации), образуя вещества АВ и К: В+АК→ В…АК →АВ+К Суммируя два последних уравнения, получим А+В=АВ т.е. в результате реакции катализатор остался без изменения.
Действие катализатор на состоянии химического равновесия не сказывается, так как катализатор в равной мере ускоряет и прямой, и обратный процесс. Катализатор ускоряет лишь достижение хим. равновесия. Каталитические реакции очень разнообразны. Во многих реакциях каталитическое влияние проявляется в скрытой форме. Сюда прежде всего относятся реакции в растворах. Как мы видели, поляризация, диссоциация и ионизация веществ в растворах – виды активации веществ – происходят под действием растворителя , к-ый, очевидно, играет в этом случае роль катализатора. Большое влияние на скорость и нпаравление процессов оказывают ионы ОН+3 и ОН-.
В зависимости от агрегатного состояния катализатора и реагирующих веществ различают катализ гомогенный и гетерогенный.Примером гомогенного катализа является реакция окисления СО (в газовой фазе в присутствии паров воды)кислородом ,а также действие разнообразных ферментов в биологических процессах.Гетерогенно-каталитическими являются процессы синтеза аммиака (катализатор железо),окисления SO2 до SO3(катализатор платина или оксид
ванадия) и т.д.
Гидролиз солей — разновидность реакций гидролиза, обусловленного протеканием реакций ионного обмена в растворах (преимущественно, водных) растворимых солей-электролитов. Движущей силой процесса является взаимодействие ионов с водой, приводящее к образованию слабого электролита в ионном или (реже) молекулярном виде («связывание ионов»).
Различают обратимый и необратимый гидролиз солей
1. Гидролиз соли слабой кислоты и сильного основания (гидролиз по аниону):
CO32− + H2O = HCO3− + OH− Na2CO3 + Н2О = NaHCO3 + NaOH (раствор имеет слабощелочную среду, реакция протекает обратимо, гидролиз по второй ступени протекает в ничтожной степени)
2. Гидролиз соли сильной кислоты и слабого основания (гидролиз по катиону):
Cu2+ + Н2О = CuOH+ + Н+ CuCl2 + Н2О = CuOHCl + HCl (раствор имеет слабокислую среду, реакция протекает обратимо, гидролиз по второй ступени протекает в ничтожной степени)
3. Гидролиз соли слабой кислоты и слабого основания:
2Al3+ + 3S2− + 6Н2О = 2Al(OH)3(осадок) + ЗН2S(газ) Al2S3 + 6H2O = 2Al(OH)3 + 3H2S (равновесие смещено в сторону продуктов, гидролиз протекает практически полностью, так как оба продукта реакции уходят из зоны реакции в виде осадка или газа).
Соль сильной кислоты и сильного основания не подвергается гидролизу, и раствор нейтрален..
Степень гидролиза
Под степенью гидролиза подразумевается отношение части соли, подвергающейся гидролизу, к общей концентрации её ионов в растворе. Обозначается α (или hгидр); α = (cгидр/cобщ)·100 % где cгидр — число молей гидролизованной соли, cобщ — общее число молей растворённой соли. Степень гидролиза соли тем выше, чем слабее кислота или основание, её образующие.
Является количественной характеристикой гидролиза.
Криоскопия (от греч. κρύο — холод и греч. σκοπέω смотрю) — метод исследования растворов, в основе которого лежит измерение понижения температуры замерзания раствора по сравнению с температурой замерзания чистого растворителя. Был предложен Ф. Раулем в 1882 году
. Криоскопия (от крио... и... скопия),метод физико-химического исследования, основанный на измерении понижения температуры замерзания раствора по сравнению с температурой замерзания чистого растворителя. Согласно Рауля законам, для бесконечно разбавленного раствора (при отсутствии электролитической диссоциации) существует зависимость Dtk = Ek×n, где Dtk — понижение температуры замерзания раствора, °С; n — концентрация раствора. Коэффициент Ek называется криоскопической постоянной растворителя. Значение Ek для различных жидкостей различно: например, для воды оно составляет 1,86, для бензола 5,07, для уксусной кислоты 3,90, для диоксана 4,63, для фенола 7,27. Зная Ek, можно вычислить молекулярную массу М вещества по формуле М=Р1×Ек·1000/Р2Dtk, гдеP1 и P2 — соответственно масса растворённого вещества и растворителя в г. Разность температур Dtк измеряют обычно метастатическим термометром или с помощью термопары. Методом К. могут быть определены значения Ekдля веществ с известной молекулярной массой, а также концентрация вещества в растворе.
Эбулиоскопия (от лат. ebulio — вскипаю) — метод исследования растворов, основанный на измерении повышения их температуры кипения по сравнению с чистым растворителем. Используется для определения молекулярной массы растворенного вещества, активности растворителя, степени диссоциации (или изотонического коэффициента).
Эбулиоскопия
Эбулиоскопия, эбуллиоскопия (от лат. ebullio — вскипаю и ...скопия), метод физико-химического исследования, основанный на измерении повышения температуры кипения раствора по сравнению с температурой кипения чистого растворителя. Согласно Рауля законам, для бесконечно разбавленного раствора нелетучего вещества и при отсутствииэлектролитической диссоциации это повышение Dtkип пропорционально моляльной концентрации т раствора: Dtkип =Еэ. m, где Еэ — коэфф. пропорциональности (эбулиоскопическая постоянная), характерный для каждого растворителя и не зависящий от природы растворённого вещества. Так, Еэ для воды равен 0,526, для бензола 2,57, для диоксана 3,27, для камфоры 6,02, для дибромэтана 6,43 (все величины выражены в °С). Э. наряду с криоскопией используется для определения молекулярной массы растворённого вещества, активностей растворителя и растворённого вещества или степени диссоциации слабого электролита. Молекулярную массу М растворённого вещества вычисляют по уравнению:
![]()
где P1 и P2— соответственно массы растворённого вещества и растворителя, выраженные в г. Разность температур Dtkип измеряют посредством метастатического термометра или чувствительной термопары.
РАВНОВЕСИЕ В СИСТЕМЕ ОСАДОК - РАСТВОР
Образование осадков
В гравиметрическом анализе необходимо получение чистых и хорошо фильтруемых осадков, т. е. крупнокри-
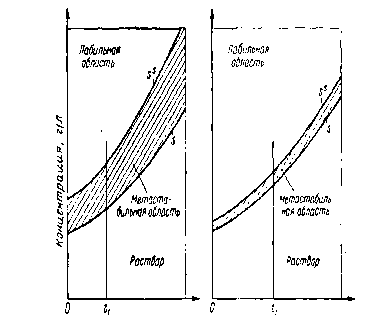
Рис. VI-1. Зависимость растворимости (5) и пересыщения (Ss) осадков от температуры для сульфата бария (а) и хлорида серебра (б):
сталлических и однороднодисперсных. Гранулометрический состав определяется относительной скоростью процессов образования центров кристаллизации и их роста.
Коэффициент формы осадка N, характеризующий число центров кристаллизации, вычисляют по уравнению:
![]()
где Р — начальная концентрация иона в растворе, S —растворимость соединения; К — константа, зависящая от растворителя и природы соединения [5, 6].
Чем меньше N, тем более крупнокристаллический осадок образуется. Можно указать следующие факторы, влияющие на значение N: разбавление растворов, температура (рис. VI-1), перемешивание, изменение рН

(рис. VI-2), комплексообразование, солевой эффект, присутствие поверхностно-активных веществ. Особенно эффективным способом получения крупнокристаллических осадков является осаждение из гомогенной системы (метод возникающих реагентов). Такое осаждение можно осуществить либо постепенным понижением растворимости, что обеспечивается постепенным повышением рН при медленно протекающей химической реакции, идущей со связыванием ионов водорода, либо постепенным увеличением концентрации ионов осади-теля или определяемого иона путем проведения реакции, протекающей с постепенным образованием этих ионов. Примером метода возникающих реагентов может служить осаждение сульфида кобальта из пиридинового раствора, осаждение диметилглиоксимата никеля синтезированным в процессе реакции осаждения реаген-том и др.
Растворимость
Растворимость, способность вещества образовывать с другим веществом однородную, термодинамически устойчивую систему переменного состава, состоящую из двух или большего числа компонентов. Такие системы возникают при взаимодействии газов с жидкостями, жидкостей с жидкостями и т.д. (см. Растворы).Соотношение компонентов может быть либо произвольным, либо ограниченным некоторыми пределами. В последнем случае Р. называют ограниченной. Мерой Р. вещества при данных условиях служит концентрация его насыщенного раствора. Р. различных веществ в определённом растворителе зависит от внешних условий, прежде всего — от температуры и давления. Давление наиболее сильно сказывается на Р. газов. Изменение внешних условий влияет на Р. в соответствии с принципом смещения равновесий (см. Ле Шателье — Брауна принцип).Для наиболее важных растворителей составлены таблицы Р. различных веществ в зависимости от внешних условий или только для стандартных условий.
Константа растворимости Ks (или произведение растворимости ПР) – произведение концентраций ионов в насыщенном растворе малорастворимого электролита – есть величина постоянная и зависит лишь от температуры.
Сольвата́ция (от лат. solvo — растворяю) — электростатическое взаимодействие между частицами (ионами, молекулами) растворенного вещества и растворителя. Сольватация в водных растворах называется гидратацией. Образующиеся в результате сольватации молекулярные агрегаты называются сольватами (в случае воды гидратами). В отличие от сольвиоза объединение однородных частиц в растворе называют ассоциацией.
Представление о сольватации ионов было введено одновременно и независимо И. А. Каблуковым и В. А. Кистяковским в 1889—1891[1].
Сольватация состоит в том, что молекула растворенного вещества оказывается окруженной сольватной оболочкой, состоящей из более или менее тесно связанных с ней молекул растворителя. В результате сольватации образуются сольваты-мол. образования постоянного или переменного состава. Время жизни сольватов определяется характером и интенсивностью межмолекулярных взаимодействий; даже в случае сильного взаимод. время жизни отдельного сольвата мало из-за непрерывного обмена частицами в сольватной оболочке. В соответствии с типами межмолекулярного взаимодействия выделяют неспецифическую и специфическую сольватацию. Неспецифическая сольватация обусловлена ван-дер-ваальсовыми взаимодействиями, специфическая сольватация проявляется главным образом вследствие электростатических взаимодействий, коор-динац. и водородных связей.
Сольватация приводит к тому, что тип растворителя изменяет скорость химических реакций (до 109 раз), определяет относительную устойчивость таутомеров, конформеров, изомеров, влияет на механизм реакций. Положения кислотно-основных равновесий в значительной степени определяются сольватирующей способностью растворителя. Подробнее о влиянии сольватации на физ.-хим, характеристики растворенных в-в и их реакц. способность см. в ст. Реакции в растворах.
На влиянии сольватации на характеристики электронных спектров поглощения и испускания основано явление, наз. сольватохромией.
Электролитическая диссоциация(ионизация)
Электролитическая диссоциация, распад вещества на ионы при растворении. Э. д. происходит вследствие взаимодействия растворённого вещества с растворителем; по данным спектроскопических методов, это взаимодействие носит в значительной мере химический характер (см. Сольватация). Наряду с сольватирующей способностью молекул растворителя определённую роль в Э. д. играет также макроскопическое свойство растворителя — его диэлектрическая проницаемость.
Классическая теория Э. д. была создана С. Аррениусом и В. Оствальдом в 80-х гг. 19 в. Она основана на предположении о неполной диссоциации растворённого вещества, характеризуемой степенью диссоциации а, т. е. долей распавшихся молекул электролита. Динамическое равновесие между недиссоциированными молекулами и ионами описывается действующих масс законом. Например, Э. д. бинарного электролита КА выражается уравнением типа КА Û К+ + А-. Константа диссоциации Кд определяется активностями катионов аК+, анионов аА- и недиссоциированных молекул аКА следующим образом:
![]() (1)
(1)
Значение Кд зависит от природы растворённого вещества и растворителя, а также от температуры и может быть определено несколькими экспериментальными методами. Степень диссоциации ее может быть рассчитана при любой концентрации a электролита с помощью соотношения:
![]() (2)
(2)
где f± — средний коэффициент активности электролита (см. также Оствальда закон разбавления).
Классическая теория Э. д. применима лишь к разбавленным растворам слабых электролитов. Сильные электролиты в разбавленных растворах диссоциированы практически полностью, поэтому представления о равновесии между ионами и недиссоциированными молекулами лишено смысла. Согласно представлениям, выдвинутым в 20—30-х гг. 20 в. В. К. Семенченко (СССР), Н. Бьеррумом (Дания), Р. М. Фуоссом (США) и др., в растворах сильных электролитов при средних и высоких концентрациях образуются ионные пары и более сложные агрегаты. Современные спектроскопические данные показывают, что ионная пара состоит из двух ионов противоположного знака, находящихся в контакте ("контактная ионная пара") или разделённых одной или несколькими молекулами растворителя ("разделённая ионная пара"). Ионные пары электрически нейтральны и не принимают участия в переносе электричества. В сравнительно разбавленных растворах сильных электролитов равновесие между отдельными сольватированными ионами и ионными парами может быть приближённо охарактеризовано, аналогично классической теории Э. д., константой диссоциации (или обратной величиной — константой ассоциации). Это позволяет использовать уравнение (2) для расчёта соответствующей степени диссоциации, исходя из экспериментальных данных.
В простейших случаях (большие одноатомные однозарядные ионы) приближённые значения константы диссоциации в разбавленных растворах сильных электролитов можно вычислить теоретически, исходя из представлений о чисто электростатическом взаимодействии между ионами в непрерывной среде — растворителе.
Степенью диссоциации (ионизации) электролита называется отношение числа молекул, распавшихся на ионы, к общему числу его молекул, введенных в ра–створ.
Иначе говоря, ан – доля молекул электролита, рас–павшихся на ионы. Степень диссоциации ан выра–жается в процентах или долях единицы:
αн = Nн/ Np,
где N – число молекул электролита, распавшихся на ионы;
Np– число молекул электролита, введенных в раст–вор (растворенных).
Так, для С(СНзСООН) = 0,1 моль/л, степень диссоциа–ции αн = 0,013 (или 1,3%).
По степени диссоциации электролиты условно по–дразделяют на сильные (αн > 30%) и слабые (αн < 3%). В промежутке электролиты считаются средней силы.
На степень электролитической диссоциации влияет коннтрация раствора.
Константа диссоциации — вид константы равновесия, которая показывает склонность большого объекта диссоциировать (разделяться) обратимым образом на маленькие объекты, как например когда комплекс распадается на составляющие молекулы, или когда соль разделяется в водном растворе на ионы. Константа диссоциации обычно обозначается Kd и обратнаконстанте ассоциации. В случае с солями, константу диссоциации иногда называют константой ионизации.
В общей реакции
![]()
где комплекс AxBy разбивается на x единиц A и y единиц B, константа диссоциации определяется так:
![]()
где [A], [B] и [AxBy] — концентрации A, B и комплекса AxBy соответственно.
Теории кислот и оснований — совокупность фундаментальных физико-химических представлений, описывающих природу и свойства кислот и оснований.
Кислотно-основные взаимодействия чрезвычайно распространенены в природе и находят широкое применение в научной и производственной практике. Теоретические представления о кислотах и основаниях имеют важное значение в формировании всех концептуальных систем химии и оказывают разностороннее влияние на развитие многих теоретических концепций во всех основных химических дисциплинах.
На основе современной теории кислот и оснований разработаны такие разделы химических наук, как химия водных и неводных растворов электролитов, рН-метрия в неводных средах, гомо- и гетерогенный кислотно-основный катализ, теория функций кислотности и многие другие.
Теория Бренстеда-Лоури
В 1923 г. Бренстед и Лоури выдвинули новую теорию кислот и оснований, основанную на представлении о переносе протона. Согласно этой теории, кислота представляет собой вещество, состоящее из молекул или ионов-доноров протонов (т. к. они отдают протоны), а основание-вещество, состоящее из молекул или ионов-акцепторов протонов (т.к. они принимают протоны).
Теория кислот и оснований Льюиса
Существуют,
однако, реакции, которые по здравому
смыслу должны относиться к кислотно-основным,
но на самом деле не подпадают под
определение Брёнстеда-Лоури. К ним
относятся, например, взаимодействия:
СаО+SO3
CaSO4
NH3+BF3NH3BF3
Для
описания реакций подобного типа Г. Льюис
(в 1923 г.) предложил новое определение
кислот и оснований. Согласно его
определению:
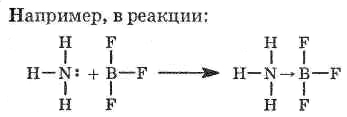 Кислотно-основная
реакция – это взаимодействие, в котором
неподелённая пара электронов молекулы
основания присоединяется к молекуле
кислоты, в результате чего возникает
ковалентная связь.
Кислотно-основная
реакция – это взаимодействие, в котором
неподелённая пара электронов молекулы
основания присоединяется к молекуле
кислоты, в результате чего возникает
ковалентная связь.
аммиак выступает в роли основания, а трифторид бора в роли кислоты. Определение основания, данное Льюисом, включает основания Брёнстеда-Лоури. Такие кислоты Льюиса, как BF3 и SO3, не являются кислотами Брёнстеда-Лоури, а такие кислоты, как НС1, H2SO4 и СН3СООН, не являются кислотами Льюиса. Теория кислот и оснований Брёнстеда-Лоури позволяет количественно определять их силу, что нельзя сказать о теории Льюиса. Следует остановиться на электронной теории кислот и оснований, предложенной Г. Льюисом. Как было отмечено выше, согласно этой теории основанием является вещество, поставляющее пару электронов для образования химической связи, а кислотой — вещество, принимающее электронную пару
Автопротолиз — гомофазный процесс самоионизации, обратимый процесс передачи протона от одной нейтральной молекулы жидкости к другой и образования в результате равного числа катионов и анионов.
Водоро́дный показа́тель, pH — мера активности (в очень разбавленных растворах она эквивалентнаконцентрации) ионов водорода в растворе, и количественно выражающая его кислотность, вычисляется как отрицательный (взятый с обратным знаком) десятичный логарифм активностиводородных ионов, выраженной в молях на литр:
![]()
Водородный показатель-определяемый как десятичный логарифм концентрации водородных ионов,взятый с обратным знаком :Hp = - lg[H+].Тогда
Нейтральная среда ……………………………………………pH=7
Кислая среда…………………………………………………...pH < 7
Щелочная среда……………………………………………….pH > 7
Сольволиз - это обменное взаимодействие растворенного вещества с растворителем, приводящее к изменению концентрации катионов и анионов растворителя. Частный случай сольволиза, когда в роли растворителя выступает вода, называется гидролизом.
Сольволиз-обменное разложение растворенного вещества и растворителя.
Буферный раствор
Буферные растворы (англ. buffer, от buff — смягчать удар) — растворы с определённой устойчивой концентрацией водородных ионов; смесь слабой кислоты и её соли (напр., СН3СООН и CH3COONa) или слабого основания и его соли (напр., NH3 и NH4CI). Величина рН буферного раствора мало изменяется при добавлении небольших количеств свободной сильной кислоты или щёлочи, при разбавлении или концентрировании. Буферные растворы широко используют в различных химических исследованиях.
Буферные растворы имеют большое значение для протекания процессов в живых организмах. Например, в крови постоянство водородного показателя рН поддерживается буферными смесями, состоящими из карбонатов и фосфатов. Известно большое число буферных растворов (ацетатно-аммиачный буферный раствор, фосфатный буферный раствор, боратный буферныйраствор, формиатный буферный раствор и др.).
Значение
pH буферного раствора можно рассчитать
по формуле: ![]() ,
где pK это отрицательный десятичный
логарифм от константы диссоциации
кислоты HA.
,
где pK это отрицательный десятичный
логарифм от константы диссоциации
кислоты HA.
Раствором называют однофазную систему переменного состава, состоящую из двух или более компонентов.Растворы представляют собой гомогенные (однородные)системы,т.е. каждый из компонентов распределен в массе другого в виде молекул,атомов или ионов .Компонент,агрегатное состояние которого не изменяется при образовании раствора, принято считать растворителем.
В случае же растворов ,образующихся при смещении газа с газом, жидкости с жидкостью ,твердого вещества с твердым, растворителем считается компонент ,количество которого в растворе преобладает.Растворы бывают газовыми ,жидкими, твердыми.
Раство́р — гомогенная (однородная) смесь, состоящая из частиц растворённого вещества, растворителя и продуктов их взаимодействия.
Истинные растворы характеризуются полной гомогенностью и благодаря небольшой разнице между размерами молекул растворенного вещества и растворителя, а также отсутствию пограничных поверхностей раздела между ними представляют собой однофазные дисперсные системы.
Для истинных растворов характерна большая прочность связи между растворенным веществом и растворителем. Растворенное вещество в дальнейшем не отделяется от раствора и, находясь под непрерывным воздействием теплового движения, остается равномерно распределенным в жидкости.
Истинные растворы — растворы, в которых частицы не могут быть обнаружены оптическим путем. Диаметр частиц в Истинных растворах меньше 10-7 см.
Коллоидными растворами называются гетерогенные дисперсные системы, в которых частицы «растворенного» вещества обладают ультрамикроскопической (коллоидной) степенью дробления. Поперечник частиц дисперсной фазы в этих системах лежит в пределах 1 -100 нм.
Даже иммерсионные микроскопы (разрешающая способность 0,2 нм) не всегда дают возможность визуально обнаружить частицы дисперсной фазы в коллоидных растворах. В то же время поперечник частиц в золях уже настолько велик (больше 7г световой волны), что свет не может свободно проходить через них и подвергается большему или меньшему рассеянию. Благодаря светорассеянию золи характеризуются феноменом Тиндаля и всегда, особенно в отраженном свете, кажутся опалесцирующими, мутноватыми или просто мутными.
В отличие от истинных растворов золи обладают очень малым осмотическим давлением, что является следствием большой относительной массы частиц.
Обратимый процесс (то есть равновесный) — термодинамический процесс, который может проходить как в прямом, так и в обратном направлении, проходя через одинаковые промежуточные состояния, причем система возвращается в исходное состояние без затрат энергии, и в окружающей среде не остается макроскопических изменений.
Обратимый процесс можно в любой момент заставить протекать в обратном направлении, изменив какую-либо независимую переменную на бесконечно малую величину.
Обратимые процессы дают наибольшую работу. Бо́льшую работу от системы вообще получить невозможно. Это придает обратимым процессам теоретическую важность. На практике обратимый процесс реализовать невозможно. Он протекает бесконечно медленно, и можно только приблизиться к нему.
Следует отметить, что термодинамическая обратимость процесса отличается от химической обратимости. Химическая обратимость характеризует направление процесса, а термодинамическая — способ его проведения.
Понятия равновесного состояния и обратимого процесса играют большую роль в термодинамике. Все количественные выводы термодинамики применимы только к равновесным состояниям и обратимым процессам\
Необратимые процессы, физические процессы, которые могут самопроизвольно протекать только в одном определённом направлении. К ним относятся: процессы диффузии, теплопроводности, термодиффузии, вязкого течения, расширения газа в пустоту и т.п. Все Н. п. являются неравновесными процессами. В замкнутых системах Н. п. сопровождаются возрастанием энтропии. В открытых системах (которые могут обмениваться энергией или веществом с окружающей средой) при Н. п. энтропия может оставаться постоянной или даже убывать за счёт обмена энтропией с внешней средой. Однако во всех случаях остаётся положительным производство энтропии, т. е. её возрастание в системе за единицу времени из-за наличия Н. п.
Классическая термодинамика, изучающая равновесные, обратимые процессы, для Н. п. устанавливает неравенства, которые указывают возможное направление Н. п. (см. Второе начало термодинамики).
Н. п. изучаются термодинамикой неравновесных процессов и статистической теорией неравновесных процессов. Термодинамика Н. п. даёт возможность находить для различных Н. п. производство энтропии в системе в зависимости от параметров неравновесного состояния, а также получать уравнения, описывающие изменения во времени этих параметров (например, уравнения диффузии, теплопроводности, Навье — Стокса уравнениегидродинамики вязкой жидкости). Коэффициенты в этих уравнениях (кинетические коэффициенты, см. Кинетика физическая) рассматриваются как феноменологические постоянные, определяемые из опыта. Статистическая теория Н. п. даёт возможность получить выражения для кинетических коэффициентов через молекулярные постоянные. Наиболее полно изучены Н. п. в газах при помощи кинетического уравнения Больцмана