

1Величину поверхности твердого тела не всегда легко измерить. Поэтому иногда скорость гетерогенной реакции относят не к единице поверхности, а к единице массы или объема твердой фазы.
Оба этих определения можно записать в математической форме. Введем обозначения: vгомог — скорость реакции в гомогенной системе; vгетерог — скорость реакции в гетерогенной системе; n — число молей какого-либо из получающихся при реакции веществ; V — объем системы; t — время; S -— площадь поверхности фазы, на которой протекает реакция; — знак приращения (n= n2-n1; = t2-t1). Тогда: vгомог=n/(St).
Первое из этих уравнений можно упростить. Отношение числа молей (n) вещества к объему (V) системы представляет собою мольно-объемную концентрацию (С) данного вещества:
n/V=C. Отсюда:
n/V=C.
И окончательно:
vгомог=C/t.
Последнее уравнение является математическим выражением другого определения скорости реакции в гомогенной системе: скоростью реакции в гомогенной системе называется изменение концентрации какого-либо из веществ, вступающих в реакцию или образующихся при реакции, происходящее в единицу времени.
Как уже говорилось, при практическом использовании химических реакций весьма важно знать, с какой скоростью будет протекать данная реакция в тех или иных условиях и как нужно изменить эти условия для того, чтобы реакция протекала с требуемой скоростью.
Скорость химической реакции зависит от природы реагирующих веществ и условий протекания реакции, важнейшими из которых являются следующие: концентрация с, температура t, присутствие катализаторов, а также от некоторых других факторов (например, от давления — для газовых реакций, от интенсивности движения жидкости или газа около поверхности, на которой происходит реакция, от измельчения — для твердых веществ, от радиоактивного излучения).
Влияние концентрации реагирующих веществ, Чтобы осуществилось химическое взаимодействие веществ А и В, их молекулы (частицы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной закон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Скорость химической реакции пропорциональна произведению реагирующих веществ.
Для гетерогенной реакции уже нельзя записать кинетическое уравнение вида:
v = k[Zn][H2SO4] (1)
Один из членов этого уравнения – «молярная концентрация цинка» - не имеет смысла, поскольку цинк находится в твердой фазе. Из опыта мы видим, что на скорость гетерогенной реакции влияет степень раздробленности твердого вещества. В принципе, этот фактор может учитываться (чисто экспериментально) константой скорости реакции – тогда для реакций с одной крупной или несколькими мелкими гранулами численные значения k будут разными, а кинетическое уравнение реакции в общем случае примет вид:
v = k[H2SO4] (2)
Итак, мы видим, что в общем случае скорость гетерогенной реакции зависит от:
а) скорости подвода реагентов к границе раздела фаз;
б) скорости реакции на поверхности раздела фаз, которая зависит от площади этой поверхности;
в) скорости отвода продуктов реакции от границы раздела фаз. Стадии (а) и (в) называются диффузионными, (см. словарь терминов) а стадия (б) – кинетической. Та стадия, которая протекает наиболее медленно, называется лимитирующей – именно она определяет скорость реакции в целом.
Универсального выражения для скорости гетерогенных реакций не существует, поскольку каждая из стадий (а-в) при определенных условиях может быть лимитирующей. Но в некоторых случаях, когда диффузионные стадии заведомо не являются лимитирующими из-за активного перемешивания реагентов, а площадь поверхности раздела фаз меняется медленно, можно экспериментально получить кинетические уравнения типа (2), удовлетворительно описывающие протекание гетерогенных реакций. Это имеет важное значение для химического производства, где большинство используемых реакций – гетерогенные.
Когда на экзамене абитуриента просят перечислить факторы, влияющие на скорость химической реакции, то обычно ожидают такого ответа:
- природа реагирующих веществ,
- концентрация реагентов,
- температура,
- наличие катализатора. Под влиянием природы реагирующих веществ обычно подразумевают то простое обстоятельство, что разные вещества реагируют по-разному. Это тривиальное наблюдение можно сделать более ценным, если анализировать примеры разной реакционной способности сходных по строению веществ в однотипных реакциях.
Что касается влияния температуры, то этот фактор действует одинаково как на скорость реакции v, так и на константу скорости k – обе эти величины быстро возрастают с повышением температуры. Полезно рассмотреть влияние температуры именно на константу скорости – в этом случае нашу задачу не осложняют постоянно меняющиеся в ходе реакции концентрации реагирующих веществ.
Еще в XIX веке голландский физикохимик Вант-Гофф опытным путем обнаружил, что при повышении температуры на 10 оС скорости многих реакций возрастают в 2-4 раза. На языке химической кинетики правило Вант-Гоффа можно выразить следующим соотношением:
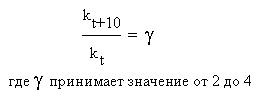
Здесь γ (греческая буква «гамма») - так называемый температурный коэффициент или коэффициент Вант-Гоффа, kt и kt+10 – константы скорости реакции при температурах t и (t + 10) оС. Для каждой конкретной реакции температурный коэффициент определяется опытным путем. Он показывает, во сколько именно раз возрастает скорость данной химической реакции (и ее константа скорости) при повышении температуры на каждые 10 градусов.
Правило Вант-Гоффа используется для приближенной оценки изменения константы скорости реакции при повышении или понижении температуры. Более точное соотношение между константой скорости и температурой установил шведский химик Сванте Аррениус:
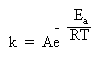
Здесь k – константа скорости, e – важное в математике число (равное приблизительно 2,71 и возникающее во многих математических преобразованиях), R – газовая постоянная, T – абсолютная температура в Кельвинах (об R и T см. в §5.6 книги 1), Ea – энергия активации. Еще одна постоянная А – «константа Аррениуса» или «предэкспонента» определяется для каждой конкретной реакции и имеет ту же размерность, что и константа скорости k.
Многие наши читатели пока не знакомы с логарифмами, поэтому здесь мы не будем приводить способ решения такого уравнения. Для нас сейчас важен общий нелинейный характер зависимости константы скорости k от температуры, а также характер связи между скоростью реакции и энергией активации. Если левую и правую части уравнения Аррениуса разделить на постоянную А и записать число e его приближенным численным значением, то уравнение примет вид:
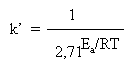
Напомним, что Ea – величина примерно постоянная для каждой реакции, не зависящая от температуры. В полученном выражении она входит в показатель степени при 2,71. Чем больше Ea конкретной реакции, тем больше будет знаменатель дроби и тем меньше (при данной температуре) будет константа скорости k (и скорость) этой реакции. Если же рассматривать какую-нибудь одну реакцию (с фиксированной Ea) при разных температурах, то повышение Т быстро уменьшает нижнюю часть дроби и, следовательно, измеряемая k будет быстро расти.
22. Ката́лиз — избирательное ускорение одного из возможных термодинамически разрешенных направлений химической реакции под действием катализатора(ов), который многократно вступает в промежуточное химическое взаимодействие с участниками реакции и восстанавливает свой химический состав после каждого цикла промежуточных химических взаимодействий.
Термин «катализ» был введён в 1835 году шведским учёным Йёнсом Якобом Берцелиусом.
Явление катализа распространено в природе (большинство процессов, происходящих в живых организмах, являются каталитическими) и широко используется в технике (в нефтепереработке и нефтехимии, в производстве серной кислоты, аммиака, азотной кислоты и др.). Большая часть всех промышленных реакций — каталитические.
Катализатор изменяет механизм реакции на энергетически более выгодный, то есть снижает энергию активации. Катализатор образует с молекулой одного из реагентов промежуточное соединение, в котором ослаблены химические связи. Это облегчает его реакцию со вторым реагентом. Важно отметить, что катализаторы ускоряют обратимые реакции, как в прямом, так и в обратном направлениях.
По влиянию на скорость реакции катализ многие источники делят на положительный (скорость реакции растет) и отрицательный (скорость реакции падает). В последнем случае происходит процесс ингибирования, который нельзя считать 'отрицательным катализом', поскольку ингибитор в ходе реакции расходуется.
Катализ бывает гомогенным и гетерогенным (контактным). В гомогенном катализе катализатор состоит в той же фазе, что и реактивы реакции, в то время, как гетерогенные катализаторы отличаются фазой.
Гомогенный катализ
Примером гомогенного катализа является разложение пероксида водорода в присутствии ионов йода. Реакция протекает в две стадии:
H2О2 + I → H2О + IO
H2О2 + IO → H2О + О2 + I
При гомогенном катализе действие катализатора связано с тем, что он вступает во взаимодействие с реагирующими веществами с образованием промежуточных соединений, это приводит к снижению энергии активации.
Гетерогенный катализ
При гетерогенном катализе ускорение процесса обычно происходит на поверхности твердого тела — катализатора, поэтому активность катализатора зависит от величины и свойств его поверхности. На практике катализатор обычно наносят на твердый пористый носитель.
Механизм гетерогенного катализа сложнее, чем у гомогенного. Механизм гетерогенного катализа включает пять стадий, причем все они обратимы.
Диффузия реагирующих веществ к поверхности твердого вещества
Физическая адсорбция на активных центрах поверхности твердого вещества реагирующих молекул и затем хемосорбция их
Химическая реакция между реагирующими молекулами
Десорбция продуктов с поверхности катализатора
Диффузия продукта с поверхности катализатора в общий поток
Примером гетерогенного катализа является окисление SO2 в SO3 на катализаторе V2O5 при производстве серной кислоты (контактный метод).
Энергия активации в химии и биологии — минимальное количество энергии, которое требуется сообщить системе (в химии выражается в джоулях на моль), чтобы произошла реакция. Термин введён Сванте Августом Аррениусом в 1889. Типичное обозначение энергии реакции Ea.
В химической модели, известной как Теория активных соударений (ТАС), есть три условия, необходимых для того, чтобы произошла реакция:
Молекулы должны столкнуться. Это важное условие, однако его не достаточно, так как при столкновении не обязательно произойдёт реакция.
Молекулы должны обладать необходимой энергией (энергией активации). В процессе химической реакции взаимодействующие молекулы должны пройти через промежуточное состояние, которое может обладать большей энергией. То есть молекулы должны преодолеть энергетический барьер; если этого не произойдёт, реакция не начнётся.
Молекулы должны быть правильно ориентированы относительно друг друга.
При низкой (для определённой реакции) температуре большинство молекул обладают энергией меньшей, чем энергия активации, и неспособны преодолеть энергетический барьер. Однако в веществе всегда найдутся отдельные молекулы, энергия которых значительно выше средней. Даже при низких температурах большинство реакций продолжают идти. Увеличение температуры позволяет увеличить долю молекул, обладающих достаточной энергией, чтобы преодолеть энергетический барьер. Таким образом повышается скорость реакции.
ГОМОГЕННЫЙ КАТАЛИЗ, ускорение хим. р-ции в присутствии катализатора, к-рый находится в одной фазе с исходными реагентами (субстратами) в газовой фазе или р-ре. При гомогенном катализе, как и при гетерогенном катализе, катализатор в р-ции не расходуется, однако является ее необходимым участником; без катализатора р-ция протекает гораздо медленнее или не идет вовсе.
Механизм гомогенно-каталитических реакций. Можно выделить сравнительно небольшую группу процессов, в к-рых участие катализатора не связано с образованием определенного хим. соед. с субстратом. К таким процессам относится, напр., катализ парамагн. частицами синглет-триплетного превращения карбенов (изменяется электронный спин молекулы) или орто-, пара-превращение Н2 (изменяется ядерный спин). Формально к гомогенному катализу можно отнести газофазные р-ции рекомбинации атомов и простейших радикалов в присут. химически инертных частиц, к-рые, участвуя в тройном соударении, обеспечивают отвод энергии, выделяющейся при образовании хим. связи. Однако в огромном большинстве случаев механизм Т.к., как и гетерог. катализа, включает более или менее сложное хим. превращ. катализатора в р-циях. В металлокомплексном гомогенном катализе р-ции ускоряются в присут. комплексных соед. Ti, Fe, Cu, Pt и др. переходных металлов, к-рые способны к образованию комплексов с молекулами субстратов. Каталитич. активность м. б. обусловлена след. факторами: 1) пространств.близостью молекул субстратов, входящих как лиганды в координац. сферу металла, 2) ослаблением хим. связей в молекулах субстратов и снижением энергии активации при их разрыве; 3) усилением вследствие электронных эффектов донорных или акцепторных св-в молекул субстратов, входящих в металлокомплекс; 4) снятием запретов по симметрии молекулярных орбиталей благодаря участию d-орбиталей металлов; 5) возможностью многоэлектронных р-ций с использованием d-электронов переходных металлов в некомплементарных окислит.-восстановит. р-циях с субстратами, для к-рых последовательное одноэлектронное окисление или восстановление термодинамически затруднено. Комплексы переходных металлов, кроме того, иногда облегчают образование своб. радикалов, что обеспечивает возможность катализа радикальных и радикально-цепных р-ций. Металлокомплексные соед. катализируют гидрирование, окисление, полимеризацию, карбонилирование олефинов и ацетиленов, фиксацию азота, диспропорционирование олефинов, активацию и разл. р-ции алканов и др.
Практическое применение. Гомогенный катализ используют для пром. получения: спиртов-гидратацией олефинов в присут. к-т (H2SO4, H3PO4); нитробензола и др. нитросоединений-нитрованием ароматич. соед. в присут. H2SO4; уксусной к-ты-карбонилированием метанола в присут. комплексов Rh (сокатализатор-HI); терефталевой к-ты-окислением п-ксилола в присут. солей Со; альдегидов - гидроформилиро-ванием олефинов в присут. карбонилов Со; лекарств, напр. L-ДОФА,-энантиоселективным гидрированием аминокислоты в присут. комплекса Rh с хиральными лигандами и др. Многие р-ции с участием металлокомплексных соед. по высокой регио- и стереоселективности приближаются к реакциям ферментативного катализа. Использование принципов действия ферментов позволяет разрабатывать принципиально новые гомогенно-каталитические процессы.
ГЕТЕРОГЕННЫЙ КАТАЛИЗ (контактный катализ), изменение скорости хим. р-ции при воздействии катализаторов, образующих самостоят. фазу и отделенных от реагирующих в-в границей раздела. наиб. распространен случай, когда твердый кат. (контакт) ускоряет р-цию между газообразными реагентами или р-цию в р-ре. Каталитич. р-ция протекает обычно на пов-сти твердого кат. и обусловлена активацией молекул реагентов при взаимод. с пов-стью. Поэтому для осуществления гетерогенного катализа необходима адсорбция компонентов реакц. смеси из объемной фазы на пов-сти катализатора.
В техн. гетерогенном катализе св-во катализатора ускорять р-цию обычно определяют как выход продукта в единицу времени, отнесенный к единице объема или массы катализатора. В теоретич. исследованиях скорость v гетерогенно-каталитич. р-ций относят к единице пов-сти катализатора и наз. удельной каталитич. активностью; ее размерность - моль х х с-1 м-2(см. Активность катализатора). Если все активные центры пов-сти однородны и равнодоступны молекулам реагирующих в-в, v пропорциональна пов-сти S Стадия гетерогенного катализа - Сложная гетерогенно-каталитич. р-ция протекает через последовательность стадий, традиционно считавшихся элементарными: подвод реагентов из объемной фазы к пов-сти катализатора (диффузия), адсорбция, хим. превращение на пов-сти (собственно катализ), десорбция продуктов, их отвод от пов-сти катализатора (диффузия). Исходными в-вами, реагирующими в элементарных р-циях, по определению, являются промежут. в-ва, к-рые образуются в предшествующих стадиях и не м. б. выделены вместе с реагирующими в-вами или продуктами р-ции. Предполагалось, что в гетерогенном катализе такими в-вами являются поверхностные соед. катализатора с реагентами, а стадии, в к-рых эти соед. образуются или распадаются,-элементарные. Развитие эксперим. методов исследования позволило установить, что в нек-рых из стадий, предполагавшихся ранее элементарными, можно выделить свои элементарные процессы, в к-рых промежут. в-ва не образуются, а изменение состояния частицы (атома, молекулы) связано с преодолением одного потенциального барьера. Рассмотрим влияние каждой из стадий на кинетич. закономерности гетерогенного катализа.
Диффузия. Если катализатор представляет собой пористые частицы (зерна), в общем случае могут иметь место след. диффузионные процессы: 1) перенос реагирующих в-в из объема газовой или жидкой фазы к внеш. пов-сти зерна катализатора (внешняя диффузия); 2) перенос частиц в порах внутри зерна (внутренняя диффузия); 3) и 4) обратные процессы - перенос частиц продуктов р-ции изнутри пор к внеш. пов-сти зерен и отвод их от внеш. пов-сти зерен в пространство между ними (соотв. внутр. и внеш. диффузия). Все эти процессы обусловлены градиентом концентраций компонентов реакц. смеси в пределах одной фазы или у границы раздела фаз. В зависимости от условий осуществления процесса (т-ры, давления, св-в катализатора) различают кинетич. область протекания гетерогенного катализа, в к-рой все диффузионные процессы происходят значительно быстрее, чем хим. р-ции, и диффузионные области (соотв. внешне- и внутренне-диффузионную), в к-рых суммарный процесс тормозится более медленным, чем хим. р-ция, переносом частиц. Поскольку с ростом т-ры и концентрации реагентов скорость хим. р-ции увеличивается быстрее, чем диффузия, диффузионное торможение в гетерогенном катализе происходит при высоких т-рах и давлениях, а также на высокодисперсных катализаторах. Катализаторы-полупроводники. Согласно электронной теории гетерогенного катализа, каталитич. активность полупроводников связана с объемной концентрацией носителей тока (электронов и дырок). Адсорбция частицы на пов-сти полупроводника приводит к образованию дополнит. (примесного) энергетич. уровня в запрещенной зоне. Переход электрона или дырки на этот уровень изменяет их объемную концентрацию и св-ва пов-сти (напр., работу выхода электрона), на к-рой возникают заряженные центры, участвующие в каталитич. превращении. Технологические особенности процессов
В пром-сти наиб. распространены реакторы с неподвижным слоем катализатора, в к-рых через слой гранулиров. или таблетированного кат. пропускается (обычно сверху вниз) поток газовых, иногда жидких, реагентов. Катализаторы, используемые в этих реакторах, кроме необходимой активности и селективности, должны обладать достаточной прочностью к истиранию, т.к. истирание увеличивает гидравлич. сопротивление слоя. Высокая уд. пов-сть и пористость катализатора повышают его общую активность, однако способствуют диффузионному торможению р-ции. Диффузионные процессы особенно вредны в случае последоват. каталитич. р-ций, когда в результате диффузионных затруднений с отводом продукта последний может претерпевать нежелательные дальнейшие превращения. Напр., при окислении этилена в этиленоксид на пористом кат. селективность может ухудшаться в результате доокис-ления С2Н4О. Для ликвидации диффузионных осложнений применяют непористые носители или дробят зерна катализатора.
Жидкофазный гетерогенный катализ проводят в реакторах смешения, в к-рых мелко зернистый кат. суспендируют в среде реагентов или р-рителя. Такие реакторы используют как в периодич., так и в непрерывном режимах. Для устранения внешне-диффузионных осложнений смесь обычно интенсивно перемешивают. По окончании р-ции катализатор необходимо отделить от реагентов.
Применяют также реакторы с кипящим, или псевдоожиженным, слоем катализатора, в к-рых пылевидный катализатор поднимается восходящим потоком жидкости или газа. Преимущества гетерогенного катализа в псевдоожиженном слое - возможность использования мелкодисперсных непористых частиц, что снижает влияние внутр. диффузии, непрерывное удаление отработанного катализатора и возможность его замены, высокий коэф. теплопередачи, позволяющий поддерживать постоянную т-ру по всему объему кипящего слоя. Псевдоожиженный слой используют для р-ций с интенсивным тепловыделением, напр. при каталитич. окислении. К его недостаткам относятся повышенная истираемость катализатора и вынос частиц катализатора из реактора, к-рые затем необходимо улавливать.
В нек-рых процессах, напр. риформинге, применяют реакторы с движущимся слоем гранулиров. катализатора, в к-рый постоянно подается свежий катализатор, а отработанный катализатор идет на регенерацию.
Для конструирования реакторов гетерогенного катализа необходимо разработать кинетич. модель процесса, к-рая позволяет определить требуемое кол-во катализатора и объем реактора, обеспечивающий макс. скорость р-ции и выход продукта. Расчеты реакторов должны учитывать также явления тепло- и массопереноса. При осуществлении экзотермич. р-ций часто используют проточно-циркуляц. схемы, включающие теплообменники между слоями катализатора. Расчеты пром. реакторов основываются на методах макрокинетики. 23. В каждом веществе запасено определенное количество энергии. С этим свойством веществ мы сталкиваемся уже за завтраком, обедом или ужином, так как продукты питания позволяют нашему организму использовать энергию самых разнообразных химических соединений, содержащихся в пище. В организме эта энергия преобразуется в движение, работу, идет на поддержание постоянной (и довольно высокой!) температуры тела.
Энергия химических соединений сосредоточена главным образом в химических связях. Чтобы разрушить связь между двумя атомами, требуется ЗАТРАТИТЬ ЭНЕРГИЮ. Когда химическая связь образуется, энергия ВЫДЕЛЯЕТСЯ.
Вспомним, что атомы не соединялись бы между собой, если бы это не вело к "выигрышу" (то есть высвобождению) энергии. Этот выигрыш может быть большим или малым, но он обязательно есть при образовании молекул из атомов.
Любая химическая реакция заключается в разрыве одних химических связей и образовании других.
Когда в результате химической реакции при образовании новых связей выделяется энергии БОЛЬШЕ, чем потребовалось для разрушения "старых" связей в исходных веществах, то избыток энергии высвобождается в виде тепла. Примером могут служить реакции горения. Например, природный газ (метан CH4) сгорает в кислороде воздуха с выделением большого количества теплоты (рис. 1-1а). Такие реакции называются ЭКЗОТЕРМИЧЕСКИМИ от латинского "экзо" - наружу (имея в виду выделяющуюся энергию).
В других случаях на разрушение связей в исходных веществах требуется энергии больше, чем может выделиться при образовании новых связей. Такие реакции происходят только при подводе энергии извне и называются ЭНДОТЕРМИЧЕСКИМИ (от латинского "эндо" - внутрь). Примером является образование оксида углерода (II) CO и водорода H2 из угля и воды, которое происходит только при нагревании. Таким образом, любая химическая реакция сопровождается выделением или поглощением энергии. Чаще всего энергия выделяется или поглощается в виде теплоты (реже - в виде световой или механической энергии). Эту теплоту можно измерить. Результат измерения выражают в килоджоулях (кДж) для одного МОЛЯ реагента или (реже) для моля продукта реакции. Такая величина называется ТЕПЛОВЫМ ЭФФЕКТОМ РЕАКЦИИ. Например, тепловой эффект реакции сгорания водорода в кислороде можно выразить любым из двух уравнений:
2 H2(г) + O2(г) = 2 H2О(ж) + 572 кДж
или
H2(г) + 1/2 O2(г) = H2О(ж) + 286 кДж
Оба уравнения одинаково правильны и оба выражают тепловой эффект экзотермической реакции образования воды из водорода и кислорода. Первое - на 1 моль использованного кислорода, а второе - на 1 моль сгоревшего водорода или на 1 моль образовавшейся воды.
Значки (г), (ж) обозначают газообразное и жидкое состояние веществ. Встречаются также обозначения (тв) или (к) - твердое, кристаллическое вещество, (водн) - растворенное в воде вещество и т.д.
Обозначение агрегатного состояния вещества имеет важное значение. Например, в реакции сгорания водорода первоначально образуется вода в виде пара (газообразное состояние), при конденсации которого может выделиться еще некоторое количество энергии. Следовательно, для образования воды в виде жидкости измеренный тепловой эффект реакции будет несколько больше, чем для образования только пара, поскольку при конденсации пара выделится еще порция теплоты.
Используется также частный случай теплового эффекта реакции - ТЕПЛОТА СГОРАНИЯ. Из самого названия видно, что теплота сгорания служит для характеристики вещества, применяемого в качестве топлива. Теплоту сгорания относят к 1 молю вещества, являющегося топливом (восстановителем в реакции окисления). Допустим, вам известна работа (в кДж), которую придется затратить для доставки ракеты с грузом с поверхности Земли до орбиты, известна также работа по преодолению сопротивления воздуха и другие затраты энергии во время полета. Как рассчитать необходимый запас водорода и кислорода, которые (в сжиженном состоянии) используются в этой ракете в качестве топлива и окислителя?
Без помощи теплового эффекта реакции образования воды из водорода и кислорода сделать это затруднительно. Ведь тепловой эффект - это и есть та самая энергия, которая должна вывести ракету на орбиту. В камерах сгорания ракеты эта теплота превращается в кинетическую энергию молекул раскаленного газа (пара), который вырывается из сопел и создает реактивную тягу.
В химической промышленности тепловые эффекты нужны для расчета количества теплоты для нагревания реакторов, в которых идут эндотермические реакции. В энергетике с помощью теплот сгорания топлива рассчитывают выработку тепловой энергии.
Врачи-диетологи используют тепловые эффекты окисления пищевых продуктов в организме для составления правильных рационов питания не только для больных, но и для здоровых людей - спортсменов, работников различных профессий. По традиции для расчетов здесь используют не джоули, а другие энергетические единицы - калории (1 кал = 4,1868 Дж). Энергетическое содержание пищи относят к какой-нибудь массе пищевых продуктов: к 1 г, к 100 г или даже к стандартной упаковке продукта. Например, на этикетке баночки со сгущенным молоком можно прочитать такую надпись: "калорийность 320 ккал/100 г".
Уравнения химических реакций, в которых вместе с реагентами и продуктами записан и тепловой эффект реакции, называются ТЕРМОХИМИЧЕСКИМИ УРАВНЕНИЯМИ.
Особенность термохимических уравнений заключается в том, что при работе с ними можно переносить формулы веществ и величины тепловых эффектов из одной части уравнения в другую. С обычными уравнениями химических реакций так поступать, как правило, нельзя.
Допускается также почленное сложение и вычитание термохимических уравнений. Это бывает нужно для определения тепловых эффектов реакций, которые трудно или невозможно измерить в опыте. Раздел химии, занимающийся изучением превращения энергии в химических реакциях, называется ТЕРМОХИМИЕЙ. Существует два важнейших закона термохимии. Первый из них, закон Лавуазье–Лапласа, формулируется следующим образом:
# Тепловой эффект прямой реакции всегда равен тепловому эффекту обратной реакции с противоположным знаком.
Это означает, что при образовании любого соединения выделяется (поглощается) столько же энергии, сколько поглощается (выделяется) при его распаде на исходные вещества. Например:
2 H2(г) + O2(г) 2 H2О(ж) + 572 кДж (горение водорода в кислороде)
2 H2О(ж) + 572 кДж = 2 H2(г) + O2(г) (разложение воды электрическим током)
Закон Лавуазье–Лапласа является следствием закона сохранения энергии.
Второй закон термохимии был сформулирован в 1840 г российским академиком Г. И. Гессом:
# Тепловой эффект реакции зависит только от начального и конечного состояния веществ и не зависит от промежуточных стадий процесса.
Это означает, что общий тепловой эффект ряда последовательных реакций будет таким же, как и у любого другого ряда реакций, если в начале и в конце этих рядов одни и те же исходные и конечные вещества.
Рассмотрим пример, поясняющий закон Гесса. Сульфат натрия Na2SO4 можно получить двумя путями из едкого натра NaOH. Один путь включает только одну стадию, а во второй - две стадии, с промежуточным получением кислой соли NaHSO4:
Первый путь (одностадийный): 2 NaOH + H2SO4 = Na2SO4 + 2 H2O + 131 кДж;
Второй путь (двухстадийный):
а) NaOH + H2SO4 = NaНSO4 + H2O + 62 кДж
б) NaHSO4 + NaOH = Na2SO4 + H2O + 69 кДж
Согласно закону Гесса, тепловой эффект получения сульфата натрия из NaOH не зависит от способа получения. Действительно, складывая тепловые эффекты двух последовательных реакций в способе (2) мы получаем тот же тепловой эффект, что и для способа (1): 65 кДж + 69 кДж = 131 кДж. Кстати, почленное сложение двух последних уравнений дает первое уравнение реакции.
Именно эти два основных закона термохимии придают термохимическим уравнениям некоторое сходство с математическими, когда в уравнениях реакций можно переносить члены из одной части в другую, почленно складывать, вычитать и сокращать формулы химических соединений. При этом необходимо учитывать коэффициенты в уравнениях реакций и не забывать о том, что складываемые, вычитаемые или сокращаемые моли вещества должны находиться в одинаковом агрегатном состоянии. Существует два основных типа энергии - кинетическая (обусловленная движением тела) и потенциальная (обусловленная положением тела или его частей в пространстве). Эти два типа энергии проявляются в различных формах, например, в виде тепловой (теплота), световой (энергия излучения), химической, электрической энергии или в других формах.
Согласно закону сохранения энергии, энергия не создается из ничего и не уничтожается, а может передаваться от одного тела к другому или превращаться из одной формы в другую. Следовательно, если в течение процесса исчезает энергия определенного вида, то взамен появляется эквивалентное количество энергии другого вида. Применим представления о сохранении энергии к химическим системам. Первое начало термодинамики — один из трёх основных законов термодинамики, представляет собой закон сохранения энергии для термодинамических систем.
Изменение внутренней энергии системы при переходе её из одного состояния в другое равно сумме работы внешних сил и количества теплоты, переданного системе, то есть, оно зависит только от начального и конечного состояния системы и не зависит от способа, которым осуществляется этот переход. Это определение особенно важно для химической термодинамики (ввиду сложности рассматриваемых процессов). Иными словами, внутренняя энергия является функцией состояния. В циклическом процессе внутренняя энергия не изменяется. Проще говоря: В любой изолированной системе запас энергии остаётся постоянным. Внутренняя энергия является функцией состояния системы, поэтому ее изменение (DU) определяется выражением:
DU = U2 – U1
Изменение внутренней энергии системы происходит при передаче энергии системе или от нее. Существует два основных способа передачи энергии - это передача теплоты и выполнение работы. Передача энергии, вызываемая разностью температур между системой и ее окружением или между одной системой и другой системой, называется передачей теплоты. Количество энергии, передаваемое таким образом, обозначается буквой Q (Дж) и равно:
Q = m·Cm·DT ,
где m - масса системы (кг),
DT - изменение температуры (K),
C - удельная теплоемкость вещества, из которого состоит система (Дж/(кг·K)).
Теплота не является свойством системы, поэтому не может быть и функцией состояния системы. Другой формой передачи энергии является работа - W (Дж). Существуют различные виды работы. В химии работа чаще всего связана с расширением системы. Такое расширение происходит при выделении газа в ходе реакции. В этом случае работа, выполняемая системой, определяется выражением:
W = p·DV = p·(V2 – V1)
где P - внешнее давление (Па), для многих химических реакций внешнее давление равно атмосферному;
Изобарный процесс — термодинамический процесс, происходящий в системе при постоянном давлении и постоянной массе идеального газа.
Согласно закону
Гей-Люссака, при изобарном
процессе в идеальном
газе ![]() .
.
Работа,
совершаемая газом при расширении или
сжатии газа, равна ![]() .
.
Количество
теплоты, получаемое или отдаваемое
газом, характеризуется изменением энтальпии: ![]() .
.
Изохорический или изохорный процесс— термодинамический процесс, который происходит при постоянном объёме. Для осуществления изохорного процесса в газе или жидкости достаточно нагревать (охлаждать) вещество в сосуде, который не изменяет своего объёма.
При изохорическом процессе давление идеального газа прямо пропорционально его температуре (см. Закон Шарля). В реальных газах закон Шарля не выполняется.
На графиках изображается линиями, которые называются изохоры. Для идеального газа они являются прямыми во всех диаграммах, которые связывают параметры: T (температура), V (объем) и P (давление). Закон Гесса — основной закон термохимии, который формулируется следующим образом:
Тепловой эффект химической реакции, проводимой в изобарно-изотермических или изохорно-изотермических условиях, зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от пути её протекания.
Иными словами, количество теплоты, выделяющееся или поглощающееся при каком-либо процессе, всегда одно и то же, независимо от того, протекает ли данное химическое превращение в одну или в несколько стадий (при условии, что температура, давление и агрегатные состояния веществ одинаковы). Например, окисление глюкозы в организме осуществляется по очень сложному многостадийному механизму, однако суммарный тепловой эффект всех стадий данного процесса равен теплоте сгорания глюкозы.
На рисунке приведено схематическое изображение некоторого обобщенного химического процесса превращения исходных веществ А1, А2… в продукты реакции В1, В2…, который может быть осуществлен различными путями в одну, две или три стадии, каждая из которых сопровождается тепловым эффектом ΔHi.
Закон открыт русским химиком Г. И. Гессом в 1840 г.; он является частным случаем первого начала термодинамики применительно к химическим реакциям. Практическое значение закона Гесса состоит в том, что он позволяет рассчитывать тепловые эффекты самых разнообразных химических процессов; для этого обычно используют ряд следствий из него. 25.
Энтропи́я (от др.-греч. ἐντροπία - поворот, превращение) — в естественных науках мера беспорядка системы, состоящей из многих элементов. В частности, в статистической физике — мера вероятности осуществления какого-либо макроскопического состояния; в теории информации — мера неопределённости какого-либо опыта (испытания), который может иметь разные исходы, а значит, и количество информации; в исторической науке, для экспликации феномена альтернативности истории (инвариантности и вариативности исторического процесса).
Энтропия в информатике — степень неполноты, неопределённости знаний.
Понятие энтропии впервые было введено Клаузиусом в термодинамике в 1865 году для определения меры необратимого рассеивания энергии, меры отклонения реального процесса от идеального. Определённая как сумма приведённых теплот, она является функцией состояния и остаётся постоянной при обратимых процессах, тогда как в необратимых — её изменение всегда положительно. Энтропия — функция состояния системы, равная в равновесном процессе количеству теплоты, сообщённой системе или отведённой от системы, отнесённому к термодинамической температуре системы.
Энтропия — функция, устанавливающая связь между макро- и микро- состояниями; единственная функция в физике, которая показывает направленность процессов. Энтропия — функция состояния системы, которая не зависит от перехода из одного состояния в другое, а зависит только от начального и конечного положения системы. Понятие энтропии как функции состояния системы постулируется вторым началом термодинамики, которое выражает через энтропию различие между необратимыми и обратимыми процессами. Для первых dS>δQ/T для вторых dS=δQ/T. Второе начало термодинамики, устанавливает существование энтропии как ф-ции состояния макроскопич. системы и вводит понятие абс. термодинамич. температуры. Утверждает, что все процессы, протекающие с конечной скоростью, в принципе необратимы, и дает термодинамич. критерии для определения направленности процессов. Вместе с первым началом термодинамики - основа классич., или феноменологич., термодинамики, которую можно рассматривать как развитую систему следствий этих двух начал. С помощью энтропии формулируются условия достижения термодинамического равновесия системы при постоянстве ее внутренней энергии, объема и числа молей i-го компонента (изолированная система) и условие устойчивости такого равновесия Это означает, что энтропия изолированной системы достигает максимума в состоянии термодинамического равновесия. Самопроизвольные процессы в системе могут протекать только в направлении возрастания энтропии.
Энтропия относится к группе термодинамических функций, называемых функциями Массье-Планка. Другие функции, принадлежащие к этой группе - функция Массье Ф1 = S - (1/T)U и фцнкция Планка Ф2 = S - (1/T)U — (p/T)V, могут быть получены в результате применения к энтропии преобразования Лежандра.
Постулат Планка (альтернативная формулировка тепловой теоремы) устанавливает, что энтропия любого химического соединения в конденсированном состоянии при абсолютном нуле температуры является условно нулевой и может быть принята за начало отсчета при определении абсолютного значения энтропии вещества при любой температуре. Уравнения (1) и (2) определяют энтропию с точностью до постоянного слагаемого.
В химической термодинамике широко используют следующие понятия: стандартная энтропия S0, т.е. энтропия при давлении р=1,01·105 Па (1 атм); стандартная энтропия химической реакции т.е. разница стандартных энтропий продуктов и реагентов; парциальная молярная энтропия компонента многокомпонентной системы dS=dQ/T Величина dS является полным дифференциалом, т.е. ее интегрирование по любому произвольно выбранному пути дает разность между значениями энтропии в начальном (А) и конечном (В) состояниях:
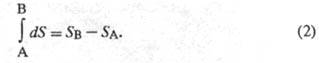
Теплота не является функцией состояния, поэтому интеграл от δQ зависит от выбранного пути перехода между состояниями А и В. Энтропия измеряется в Дж/(моль·град).
26.
Свободная энергия Гиббса (или просто энергия Гиббса, или потенциал Гиббса, или термодинамический потенциал в узком смысле) — это величина, показывающая изменение энергии в ходе химической реакции и дающая таким образом ответ на вопрос о принципиальной возможности протекания химической реакции; это термодинамический потенциал следующего вида:
G=U+PV-TS
Энергию Гиббса можно понимать как полную химическую энергию системы (кристалла, жидкости и т. д.)
Понятие энергии Гиббса широко используется в термодинамике и химии.
Самопроизвольное протекание изобарно-изотермического процесса определяется двумя факторами: энтальпийным, связанным с уменьшением энтальпии системы (ΔH), и энтропийным T ΔS, обусловленным увеличением беспорядка в системе вследствие роста ее энтропии. Разность этих термодинамических факторов является функцией состояния системы, называемой изобарно-изотермическим потенциалом или свободной энергией Гиббса (G, кДж) Классическим определением энергии Гиббса является выражение
![]()
где ![]() — внутренняя
энергия,
— внутренняя
энергия, ![]() — давление,
— давление, ![]() — объем,
— объем, ![]() —
абсолютная температура,
—
абсолютная температура, ![]() — энтропия.
— энтропия.
В
химических процессах одновременно
действуют два противоположных
фактора — энтропийный (![]() )
и энтальпийный (
)
и энтальпийный (![]() ).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (
).
Суммарный эффект этих противоположных
факторов в процессах, протекающих при
постоянном давлении и температуре,
определяет изменение энергии
Гиббса (![]() ):
):
![]()
Из
этого выражения следует, что ![]() ,
то есть некоторое количество
теплоты расходуется
на увеличение энтропии (
),
эта часть энергии потеряна для совершения
полезной работы,
её часто называют связанной
энергией.
Другая часть теплоты (
,
то есть некоторое количество
теплоты расходуется
на увеличение энтропии (
),
эта часть энергии потеряна для совершения
полезной работы,
её часто называют связанной
энергией.
Другая часть теплоты (![]() )
может быть использована для совершения
работы, поэтому энергию Гиббса часто
называют такжесвободной
энергией.
)
может быть использована для совершения
работы, поэтому энергию Гиббса часто
называют такжесвободной
энергией.
Характер
изменения энергии Гиббса позволяет
судить о принципиальной возможности
осуществления процесса. При ![]() процесс
может протекать, при
процесс
может протекать, при ![]() процесс
протекать не может (иными словами, если
энергия Гиббса в исходном состоянии
системы больше, чем в конечном, то процесс
принципиально может протекать, если
наоборот — то не может). Если же
процесс
протекать не может (иными словами, если
энергия Гиббса в исходном состоянии
системы больше, чем в конечном, то процесс
принципиально может протекать, если
наоборот — то не может). Если же ![]() ,
то система находится в состоянии химического
равновесия.
,
то система находится в состоянии химического
равновесия.
Обратите внимание, что речь идёт исключительно о принципиальной возможности протекания реакции. В реальных же условиях реакция может не начинаться и при соблюдении неравенства (по кинетическим причинам).
Существует
полезное соотношение, связывающее
изменение свободной энергии Гиббса
в
ходе химической реакции с её константой
равновесия ![]() :
:
![]()
Вообще говоря, любая реакция может быть рассмотрена как обратимая (даже если на практике она таковой не является). При этом константа равновесия определяется как
![]()
где ![]() — константа
скорости прямой
реакции,
— константа
скорости прямой
реакции, ![]() —
константа скорости обратной реакции.
—
константа скорости обратной реакции.
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1885 году французским ученым Ле-Шателье.
Факторы влияющие на химическое равновесие:
1) температура
При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции.
CaCO3=CaO+CO2 -Q t↑ →, t↓ ←
N2+3H2↔2NH3 +Q t↑ ←, t↓ →
2) давление
При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся.
CaCO3=CaO+CO2 P↑ ←, P↓ →
1моль=1моль+1моль
3) концентрация исходных веществ и продуктов реакции
При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при увеличении концентрации продуктов реакции-в сторону исходных веществ.
S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ←
Катализаторы не влияют на смещение химического равновесия! Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции.
Для реакции в смеси идеальных газов константа равновесия может быть выражена через равновесные парциальные давления компонентов pi по формуле[1]:
![]()
где νi — стехиометрический коэффициент (для исходных веществ принимается отрицательным, для продуктов — положительным). Kp не зависит от общего давления, от исходных количеств веществ или от того, какие участники реакции были взяты в качестве исходных, но зависит от температуры [2].
Например, для реакции окисления монооксида углерода:
2CO + O2 = 2CO2
константа равновесия может быть рассчитана по уравнению:
![]()
Если реакция протекает в идеальном растворе и концентрация компонентов выражена через молярность ci, константа равновесия принимает вид:
![]()
Для реакций в смеси реальных газов или в реальном растворе вместо парциального давления и концентрации используют соответственно фугитивность fi и активность ai:
![]()
![]()
В
некоторых случаях (в зависимости от
способа выражения) константа равновесия
может являться функцией не только
температуры, но и давления. Так, для
реакции в смеси идеальных газов
парциальное давление компонента может
быть выражено по закону
Дальтона через
суммарное давление и мольную долю
компонента (![]() ),
тогда легко показать[2],
что:
),
тогда легко показать[2],
что:
![]()
где Δn —
изменение числа молей веществ в ходе
реакции. Видно, что Kx зависит
от давления. Если число молей продуктов
реакции равно числу молей исходных
веществ (![]() ),
то
),
то ![]() .
.
Для обратимой химической реакции константа равновесия может быть выражена через константы скорости прямых и обратных реакций, исходя из того факта, что в состоянии равновесия скорости прямой и обратной реакций равны. Например, для элементарной обратимой химической реакции первого порядка
![]()
легко показать[2], что:
![]()
где k1 — константа скорости прямой реакции, а k2 — обратной. Это важное соотношение даёт одну из «точек соприкосновения» химической кинетики и химической термодинамики.
Энтропийный метод расчёта ΔG реакции является одним из самых распространённых и удобных[2]. Он основан на соотношении:
![]()
или, соответственно, для стандартного изменения энергии Гиббса:
![]()
27.
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причём скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.
Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Влияние, которое оказывают эти факторы на химическую реакцию, подчиняются закономерности, которая была высказана в общем виде в 1885 году французским ученым Ле-Шателье.
Факторы влияющие на химическое равновесие:
1) температура
При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции.
CaCO3=CaO+CO2 -Q t↑ →, t↓ ←
N2+3H2↔2NH3 +Q t↑ ←, t↓ →
2) давление
При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся.
CaCO3=CaO+CO2 P↑ ←, P↓ →
1моль=1моль+1моль
3) концентрация исходных веществ и продуктов реакции
При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при увеличении концентрации продуктов реакции-в сторону исходных веществ.
S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ←
Катализаторы не влияют на смещение химического равновесия!
Принцип Ле Шателье — Брауна (1884 г.) — если на систему, находящуюся в устойчивом равновесии, воздействовать извне, изменяя какое-либо из условий равновесия (температура, давление, концентрация, внешнее электромагнитное поле), то в системе усиливаются процессы, направленные на компенсацию внешнего воздействия.
Анри Ле Шателье (Франция) сформулировал этот термодинамический принцип подвижного равновесия, позже обобщённый Карлом Брауном. Влияние температуры зависит от знака теплового эффекта реакции. При повышении температуры химическое равновесие смещается в направлении эндотермической реакции, при понижении температуры — в направлении экзотермической реакции. В общем же случае при изменении температуры химическое равновесие смещается в сторону процесса, знак изменения энтропии в котором совпадает со знаком изменения температуры.
Давление существенно влияет на положение равновесия в реакциях с участием газообразных веществ, сопровождающихся изменением объёма за счёт изменения количества вещества при переходе от исходных веществ к продуктам:
При повышении давления равновесие сдвигается в направлении, в котором уменьшается суммарное количество молей газов и наоборот.
В реакции синтеза аммиака количество газов уменьшается вдвое: N2 + 3H2 ↔ 2NH3
Значит, при повышении давления равновесие смещается в сторону образования NH3, о чем свидетельствуют следующие данные для реакции синтеза аммиака при 400 °C.
Введение в реакционную смесь или образование в ходе реакции инертных газов действует так же, как и понижение давления, поскольку понижается парциальное давление реагирующих веществ. Следует отметить, что в данном случае в качестве инертного газа рассматривается газ, не участвующий в реакции. В системах с уменьшением количества молей газов инертные газы смещают равновесие в сторону исходных веществ, поэтому в производственных процессах, в которых могут образовываться или накапливаться инертные газы, требуется периодическая продувка газоводов.
Влияние концентрации на состояние равновесия подчиняется следующим правилам:
При повышении концентрации одного из исходных веществ равновесие сдвигается в направлении образования продуктов реакции;
При повышении концентрации одного из продуктов реакции равновесие сдвигается в направлении образования исходных веществ.
Конста́нта равнове́сия — величина, определяющая для данной химической реакции соотношение между термодинамическими активностями (либо, в зависимости от условий протекания реакции, парциальными давлениями, концентрациями или фугитивностями) исходных веществ и продуктов в состоянии химического равновесия (в соответствии с законом действующих масс). Зная константу равновесия реакции, можно рассчитать равновесный состав реагирующей смеси, предельный выход продуктов, определить направление протекания реакции. Подробнее про КС смотри в предыдущем билете. 28. Повторение предыдущего.