

38. Вывести уравнение Аррениуса. Энергия активации. Предэкспоненциальный множитель. Методы их определения.
Рассмотрим термодинамический вывод выражения, описывающего зависимость константы скорости реакции от температуры и величины энергии активации – уравнения Аррениуса. Согласно уравнению изобары Вант-Гоффа,
![]() (II.31)
(II.31)
Поскольку константа равновесия есть отношение констант скоростей прямой и обратной реакции, можно переписать выражение (II.31) следующим образом:
![]() (II.32)
(II.32)
Представив изменение энтальпии реакции ΔHº в виде разности двух величин E1 и E2, получаем:
![]() (II.33)
(II.33)
![]() (II.34)
(II.34)
Здесь С – некоторая константа. Постулировав, что С = 0, получаем уравнение Аррениуса, где EA – энергия активации:
![]() (II.35)
(II.35)
После неопределенного интегрирования выражения (II.35) получим уравнение Аррениуса в интегральной форме:
![]() (II.36)
(II.36)
![]() (II.37)
(II.37)
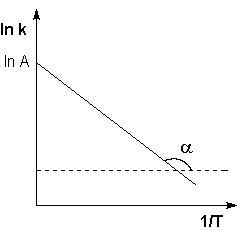
Рис. 2.7 Зависимость логарифма константы скорости химической
реакции от обратной температуры.
Здесь A – постоянная интегрирования. Из уравнения (II.37) нетрудно показать физический смысл предэкспоненциального множителя A, который равен константе скорости реакции при температуре, стремящейся к бесконечности. Как видно из выражения (II.36), логарифм константы скорости линейно зависит от обратной температуры (рис.2.7); величину энергии активации EA и логарифм предэкспоненциального множителя A можно определить графически (тангенс угла наклона прямой к оси абсцисс и отрезок, отсекаемый прямой на оси ординат).
![]() (II.38)
(II.38)
Зная энергию активации реакции и константу скорости при какой-либо температуре T1, по уравнению Аррениуса можно рассчитать величину константы скорости при любой температуре T2:
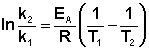
Энергия активации (E) – тот избыток энергии по сравнению со средней энергии молекул при данной температуре, которой должны обладать молекулы, чтобы они могли вступить в хим. реакцию. Если E↑, то υ↓
![]() ур-ние Аррениуса
ур-ние Аррениуса
графический:
![]()
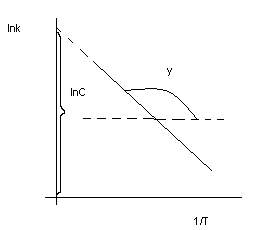
lnC – константа интегрирования
![]()
Отсюда определяем величину энергии активации.
по энергетическому барьеру:
и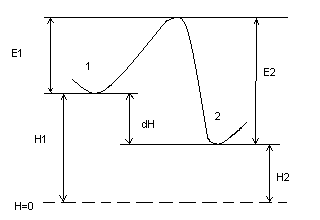 з
состояния 1 в состояние 2 при затрате
энергии Е1, обратный переход при
затрате энергии Е2
з
состояния 1 в состояние 2 при затрате
энергии Е1, обратный переход при
затрате энергии Е2
∆Н = Е2 - Е1
-∆Н = Н1 - Н2
Константа скорости не зависит от концентрации и зависит только от температуры. Эта зависимость называется законом Аррениуса (1889) и, в соответствии со вторым постулатом, имеет вид:
(1.5)
Здесь R – универсальная газовая постоянная и ko – предэкспоненциальный фактор, пропорциональный числу столкновений молекул с подходящей пространственной ориентацией, а множитель учитывает долю молекул, имеющих при столкновении энергию Е ≥ Ea (закон Больцмана).
39. Порядок реакции по реагенту и методы его экспериментального определения. Лимитирующая стадия химического процесса.
Для определения порядка реакции по реагенту известно несколько методов, которые излагаются ниже. В этом случае для точного определения порядка реакции по реагенту А все эксперименты проводятся при постоянной температуре и концентрации прочих реагентов (если имеются).
Наиболее просто можно определить порядок реакции из кинетической кривой СА(t), типа изображенной на рис. 1.
С C (0) С1 А С2 N M
|
Рис. 1. Изменение концентрации реагента во времени реакции: C(0), С1 и С2 начальная и текущая концентрация реагента в моменты t1 и t2
|
t1 t2 Время |
В различные моменты времени, при соответствующей концентрации определяют скорость реакции путем дифференцирования кривой (графически или аналитически), получая набор значений r(СА). Например, порядок реакции 1 по реагенту A можно найти из выражения:
log ri = log k. + а1.log CAi (1.19)
представленного в виде графика «lg
ri– lgCАi»,
причем тангенс угла наклона прямой в
этих координатах численно равен порядку
реакции mА. Найденный
из кинетической кривой порядок реакции
называют
![]() .
.
В методе Вант-Гоффа проводится серия экспериментов с различными начальными концентрациями реагента А, в которых определяется начальная скорость реакции, r0(С(0)А). Затем также строят график в координатах «lgr0–lgCА» и находят порядок реакции как тангенс угла наклона прямой в указанных координатах. Найденный таким образом порядок реакции называют концентрационным. Для элементарных реакций оба метода дают одинаковый результат; в случае сложных реакций получаются различные значения временного и концентрационного порядка реакции.
lg r0
--------
|
Рис. 7. Определение порядка реакции по экспериментальным данным: tg = mА |



 lgC(0)A
lgC(0)A
Например, для газофазной реакции: Н2 + Br2 2 НBr концентрационный порядок по брому равен 0,5, а временной порядок по мере расхода брома увеличивается от 0,5 до 1,5 (при завершении реакции в замкнутой системе).
Можно определить порядок реакции по зависимости времени полупревращения от начальной концентрации реагента. Ниже будет показано, что для элементарных реакций эти величины взаимосвязаны (см. таблицу кинетических уравнений).
Порядок реакции можно найти, анализируя применимость соответствующих кинетических уравнений для описания кривых С(t) в линеаризующих координатах (см. ниже).
Для определения порядка реакции может быть полезным метод Оствальда- понижение порядка на единицу путем применения большого избытка одного из реагентов.
В стационарном состоянии системы можно использовать допущение о лимитирующей стадии, т.е. предположение о том, что скорость суммарного процесса определяется скоростью наиболее медленной стадии этого процесса, например, стадия 2 для схемы R9. Тогда скорость этой стадии равна:
r2 = k2 [B] = k2K1[A] (2.27)
в этом случае
b = K1 exp(-K1k2t) (2.28)
d = 1- (1+K1)exp(-K1k2t) (2.29)
Следует отметить (аналогия с гидравликой), что до лимитирующей стадии происходит накопление вещества, а после ЛС все концентрации промежуточных соединений весьма малы. При наличии ЛС вся кинетическая информация относится только к этой медленной стадии. В сложном многостадийном процессе при наличии ЛС всегда реализуется стационарный режим. Действительно, в таком случае концентрации реагентов изменяются со скоростью медленной ЛС, а концентрации промежуточных соединений практически равновесны.