

Гликолиз на начальном этапе гипоксии активируется.
Причины:
Дефицит АТФ и снижение его ингибирующего влияния на ключевые ферменты гликолиза.
Активация гликолитических ферментов продуктами гидролиза АТФ: АДФ и АМФ.
Проявления:
Снижение содержания гликогена и глюкозы в клетках.
Увеличение внутриклеточного содержания молочной и пировиноградной кислот.
Последнее является также результатом торможения их окисления в дыхательной цепи и ресинтеза из них гликогена, требующего энергии АТФ.
Содержание Н+ в клетках и биологических жидкостях прогрессирующе нарастает и развивается ацидоз вследствие торможения окисления субстратов, особенно - лактата и пирувата, КТ и в меньшей мере - жирных кислот и аминокислот.
Биосинтез нуклеиновых кислот и белков подавлен вследствие дефицита энергии, необходимой для этих процессов.
Параллельно активируется протеолиз, обусловленный активацией в условиях ацидоза протеаз, а также неферментного гидролиза белков.
Азотистый баланс становится отрицательным. Это сочетается с повышением уровня остаточного азота в плазме крови и аммиака в тканях. Причины: активация реакций протеолиза и торможение процессов протеосинтеза.
Жировой обмен характеризуется:
Активацией липолиза вследствие повышения активности липаз и ацидоза.
Торможением ресинтеза липидов. Причина: дефицит макроэргических соединений.
Накоплением в результате вышеуказанных процессов избытка КТ (ацетоуксусной, β-оксимасляной кислот, ацетона) и жирных кислот в плазме крови, межклеточной жидкости, клетках. При этом ВЖК оказывают разобщающее влияние на процессы окисления и фосфорилирования, что усугубляет дефицит АТФ.
Обмен электролитов и жидкости в тканях существенно нарушен.
Причины:
Дефицит АТФ, энергия которой необходима для АТФаз: Nа+,К+-АТФазы, Са2+-зависимой АТФазы и др.
Повреждение мембран и их ионных каналов, обеспечивающих энерго- и электрозависимый перенос, а также пассивный транспорт ионов.
Изменение содержания в организме гормонов, регулирующих обмен ионов: минералокортикоидов, кальцитонина и др.
Проявления:
Нарушение соотношения ионов в клетках:
Трансмембранного (обычно в условиях гипоксии клетки теряют К+, в цитозоле накапливаются Na+ и Са2+, в митохондриях - Са2+).
Между отдельными ионами (например, в цитозоле уменьшается соотношение K+/Na+, К+/Са2+).
Увеличение в крови содержания Na+, C1-, отдельных микроэлементов.
Накопление избытка жидкости в клетках (набухание клеток).
Причины:
Увеличение осмотического давления в цитоплазме клеток в связи с накоплением в них Na+, Са2+ и некоторых других ионов, а также в результате гидролиза крупных молекул органических веществ (например, гликогена, белка).
Повышение онкотического давления в клетках в результате распада полипептидов, ЛП и других белоксодержащих молекул, обладающих гидрофильными свойствами.
При гипоксии нарушения функций органов и тканей выражены в разной мере, Это определяется различной резистентностью органов к гипоксии, а также скоростью её развития, степенью и длительностью её воздействия на организм.
Патогенетическая классификация гипоксий (по и.Р. Петрову)
Экзогенный тип гипоксии или гипоксическая гипоксия - гипоксия вследствие снижения парциального давления кислорода во вдыхаемом воздухе (характерно снижение артерио-венозной разницы по кислороду).
Гипобарическая форма возникает при снижении общего барометрического давления (подъем на высоту).
Нормобарическая форма возникает при избирательном снижении содержания кислорода при нормальном общем давлении (нахождение в замкнутых или плохо проветриваемых пространствах).
Гипоксия при патологических процессах, нарушающих снабжение тканей кислородом при нормальном содержании его в окружающей среде или утилизации кислорода из крови при нормальном ее насыщении О2.
Дыхательный (респираторный) тип гипоксии возникает при недостаточном транспорте кислорода из нормального атмосферного воздуха в плазму протекающей через легкие крови вследствие нарушения системы внешнего дыхания (характерна артериальная гипоксемия).
Сердечно-сосудистый (циркуляторный) тип гипоксии возникает в результате нарушений гемодинамики, приводящих к недостаточному для нормальной жизнедеятельности снабжению органов и тканей кислородом при нормальном насыщении им артериальной крови (характерна венозная гипоксемия).
Кровяной (гемический) тип гипоксии возникает в результате неспособности крови при наличии нормального напряжения кислорода в легочных капиллярах связывать, переносить в ткани и отдавать нормальное количество кислорода, т. е. патогенетической основой данного типа гипоксии является уменьшение реальной кислородной емкости крови (характерна артериальная гипоксемия).
Тканевой (гистотоксический) тип гипоксии возникает в результате нарушения процессов биологического окисления в клетках при нормальном функционировании всех звеньев системы транспорта кислорода к месту его утилизации (характерно снижение артерио-венозной разницы по кислороду).
Смешанный тип гипоксии.
Один и тот же фактор вызывает сочетание двух или более типов гипоксии.
Первично возникает один тип гипоксии, а затем по мере развития болезни присоединяются другие типы.
Физиологическая (перегрузочная) гипоксия – возникает при физических нагрузках, особенно тяжелых, обусловлена появлением кислородного долга (характерны венозная гипоксемия, гиперкапния и увеличение артерио-венозной разницы по кислороду).
Срочная адаптация к гипоксии (или 1-ая стадия долговременной адаптации)
Приспособительные реакции системы внешнего дыхания:
увеличение альвеолярной вентиляции за счет углубления и учащения дыхания и мобилизации резервных альвеол (вызывает развитие дыхательного алкалоза, кривая диссоциации HbО2 сдвигается влево и оксигенация крови улучшается);
увеличение легочного кровотока и повышение перфузионного давления в капиллярах легких;
возрастание проницаемости альвео-капиллярных мембран для газов.
Приспособительные реакции в системе кровообращения:
развитие тахикардии, увеличение ударного и минутного объемов сердца;
увеличение массы циркулирующей крови за счет выброса из кровяного депо;
увеличение системного артериального давления и скорости кровотока;
расширение сосудов (под влиянием СО2, рН, аденозина).
Приспособительные реакции системы крови:
усиление диссоциации оксиHb за счет ацидоза;
повышение кислородной емкости крови за счет усиления вымывания эритроцитов из костного мозга;
Тканевые приспособительные реакции:
ограничение функциональной активности органов и тканей, непосредственно не участвующих в обеспечении транспорта кислорода;
увеличение сопряжения окисления и фосфорилирования и активности ферментов дыхательной цепи;
усиление анаэробного синтеза АТФ за счет активации гликолиза (накапливается лактат, рН смещается в кислую сторону, а кривая Баркрофта – вправо, HbО2 легче диссоциирует, отдавая кислород).
Стадия срочной адаптации может развиваться по двум направлениям:
Если действие гипоксического фактора прекращается, то адаптация не развивается и функциональная система ответственная за адаптацию к гипоксии не закрепляется.
Если действие гипоксического фактора продолжается или периодически повторяется в течение достаточно длительного времени, то организм переходит во 2-ю стадию долгосрочной адаптации.
2-я стадия - переходная
Ей характерно постепенное снижение активности систем, обеспечивающих приспособление организма к гипоксии, и ослабление стрессовых реакций на повторное действие гипоксического фактора.
3-я стадия - стадия устойчивой долговременной адаптации
Она характеризуется высокой резистентностью организма к гипоксическому фактору.
увеличение силы дыхательных мышц и дыхательной поверхности легких;
гипертрофия миокарда;
активация эритропоэза за счет усиления образования эритропоэтинов в почках и, возможно, других органах;
увеличение массы митохондрий.
4-я стадия
Если действие гипоксического фактора прекращается, то постепенно происходит дезадаптация организма.
Если действие гипоксического фактора нарастает, то это может привести к истощению функциональной системы и произойдет срыв адаптации и полное истощение организма.
18. – Этиология и патогенез циркуляторной гипоксии
Сердечно-сосудистый (циркуляторный) тип гипоксии возникает в результате нарушений гемодинамики, приводящих к недостаточному для нормальной жизнедеятельности снабжению органов и тканей кислородом при нормальном насыщении им артериальной крови (характерна венозная гипоксемия).
Главным гемодинамическим показателем, характеризующим циркуляторную гипоксию, является уменьшение по сравнению с должными величинами скорости кровотока (Q), т. е. количества крови, протекающей через суммарный просвет микрососудов в единицу времени. Q зависит от нескольких факторов:
Q = f (V, ‑P, W, R), где:
V - объем крови, циркулирующий в участке ткани, органе или организме в целом.
‑P = Pa - Pв - градиент давления между артериальным отделом русла (Ра) и венозным (Рв).
W - суммарный тонус сосудов данного бассейна.
R - реологические свойства крови.
Таким образом, развитие данного типа гипоксии может быть обусловлено любым из перечисленных гемодинамических факторов и изменениями текучести крови. Часто имеет место сочетание двух или более факторов.
Коррекция циркуляторных гипоксий Коррекция циркуляторных гипоксий местного характера частично описана в разделе "Патология микроциркуляции".
Общие или генерализованные циркуляторные гипоксии возникают по трем кардинальным причинам и требуют, соответственно, трех различных подходов.
Ослабление сердечной деятельности в зависимости от характера сердечной недостаточности (острой или хронической) требует назначения кардиотонических средств либо быстрого действия (обычно адреналина или других препаратов с β1-адреномиметическим действием, а также блокаторы фосфодиэстеразы – амринон, милринон); либо более медленнодействующих сердечных гликозидов (строфантин, дигоксин).
Снижение системного сосудистого тонуса при коллапсах, шоках корректируется средствами, возбуждающими сосудодвигательный центр (уже упоминавшиеся аналептики), α-адренорецепторы адреналин, норадреналин).
Снижение объема циркулирующей крови требует назначения плазмозамещающих растворов (полиглюкин, реополиглюкин ).
19. – Этиология и патогенез респираторной гипоксии
Дыхательный (респираторный) тип гипоксии возникает при недостаточном транспорте кислорода из нормального атмосферного воздуха в плазму протекающей через легкие крови вследствие нарушения системы внешнего дыхания (характерна артериальная гипоксемия).
Механизмы развития:
Альвеолярная гиповентиляция.
Нарушение общей легочной перфузии.
Локальные нарушения вентиляционно-перфузионных отношений.
Избыточное шунтирование венозной крови в легких.
Затруднение диффузии кислорода через альвео-капиллярную мембрану.
Коррекция дыхательной гипоксии
Если гипоксия зависит от угнетения дыхательного центра, применяют препараты, прямо или косвенно возбуждающие дыхательный центр, так называемые дыхательные аналептики ("оживляющие").
Прямое стимулирующее действие оказывают препараты "стволового" действия, то есть, стимулирующие стволовую часть головного мозга, включающую и продолговатый мозг с его жизненно важными центрами, в том числе – дыхательным (коразол, кордиамин, бемегрид, камфора).
Широко применяют препараты, рефлекторно возбуждающие дыхательный центр.
N-холиномиметики, аналоги никотина, активирующие рецепторы дуги аорты и каротидного синуса, связанные афферентными волокнами с дыхательным центром (цититон, лобелин). В силу высокой токсичности для центральной нервной системы (как известно, тот же никотин вслед за стимуляцией оказывает блокирующее действие на N-холинорецепторы), данные препараты вводятся в небольшой дозе, но только внутривенно, для кратковременного создания высокой концентрации вокруг рецепторов указанных зон, поэтому в дальнейшем, разбавившись в крови, они уже не в состоянии оказать токсического действия на центральную нервную систему.
Ингаляции газовой смеси с повышенной концентрацией естественного стимулятора - углекислого газа (карбоген).
Если нарушение внешнего дыхания произошло из-за изменений бронхиальной проходимости (чаще всего бронхиальная астма), применяют весь набор препаратов, расширяющих гладкие мышцы и снижающих отек слизистых бронхов (стимуляторы аденилатциклазы или блокаторы фосфодиэстеразы адреномиметики, диметилксантины; а также М-холиноблокаторы, спазмолитики, блокаторы выхода гистамина из тучных клеток, блокаторы Н1 гистаминовых рецепторов, блокаторы брадикининовых рецепторов, блокаторы синтеза антител и другие иммуносупрессоры). Подробно эти подходы рассматриваются в разделе “Патология внешнего дыхания”.
Дыхательная недостаточность, вызванная повреждением дыхательной мускулатуры (миастения) или снижением функции диафрагмального нерва поддается коррекции путем назначения препаратов, стимулирующих N-холинорецепторы, но уже скелетных мышц. Прямая стимуляция N-холиномиметиками из-за уже упомянутой высокой токсичности недопустима, поэтому применяют обычно антихолинэстеразные препараты (физостигмин, прозерин, галантамин).
Нарушения дыхания из-за гидроторакса требуют в большинстве случаев назначения
мочегонных препаратов быстрого действия (фуросемид, этакриновая кислота),
препаратов, устраняющих причину гидроторакса (противомикробных и противовоспалительных при пневмонии, кардиотонических при сердечных водянках и так далее).
Иногда дыхательная гипоксия возможна и ятрогенного генеза вследствие передозировки во время операции миорелаксантов – препаратов, блокирующих N-холинорецепторы мышц. Остановка дыхания вследствие применения миорелаксантов деполяризующего действия (сходных по действию с ацетилхолином) не поддается фармакологической коррекции, помощь заключается только в поддержании искусственной вентиляции на время, пока не произойдет разрушения препарата холинэстеразой. Передозировка антидеполяризующими миорелаксантами (препятствующими действию эндогенного ацетилхолина), ликвидируется антихолинэстеразными веществами, когда накапливающийся эндогенный ацетилхолин постепенно вытесняет миорелаксант из связи с рецептором.
20. – Этиология и патогенез гемической гипоксии
Кровяной (гемический) тип гипоксии возникает в результате неспособности крови при наличии нормального напряжения кислорода в легочных капиллярах связывать, переносить в ткани и отдавать нормальное количество кислорода, т. е. патогенетической основой данного типа гипоксии является уменьшение реальной кислородной емкости крови (характерна артериальная гипоксемия).
Причины развития:
Уменьшение количества гемоглобина.
Качественные изменения гемоглобина наследственного и приобретенного генеза.
Нарушения физико-химических условий, необходимых для нормального поглощения кислорода гемоглобином из плазмы крови легочных капилляров и отдачи кислорода в тканевых капиллярах.
Коррекция гемических гипоксий
Гемические гипоксии корректируются с помощью фармакотерапии анемии. Поддаются коррекции железодефицитные анемии (препараты железа, препараты, улучшающие всасывание и ионизацию его, препараты, направленные на борьбу с причинами железодефицитных состояний, например, усиление гемостаза при постгеморрагических анемиях), применяется иммуносупрессия при гемолитических анемиях аутоиммунного генеза). Другие варианты коррекции анемий представлены в теме "Патология красной крови". Восстановление инактивированного гемоглобина (карбокси- или метгемоглобина) пока недостаточно хорошо поддается фармакокоррекции и требует либо замены эритроцитов, либо повышения той фракции кислорода, которая растворена в плазме с помощью назначения кислорода, а лучше - гипербарической оксигенции.
Следует упомянуть о возможности ятрогенного фармакотерапевтического образования метгемоглобина при передозировке, например, препаратов – производных анилина (парацетамол).
21. – Боль. Виды. Механизмы формирования патологической боли. Рецепторы, проводники, нейроны, медиаторы.
Боль – субъективное неприятное ощущение, вызванное угрозой повреждения или повреждением тканей, сопровождающееся изменением двигательной, вегетативной и эмоциональной сфер организма для защиты от повреждения.
Боль - неприятное ощущение и эмоциональное переживание, возникающее в связи с настоящей или потенциальной угрозой повреждения тканей или изображаемой терминами такого повреждения (H. Merskey and N. Bogduk).
Боль – «сторожевой пес здоровья».
Боль – типовой адаптивно-приспособительный и патологический процесс.
Боль всегда субъективна. Каждая личность воспринимает и применяет это слово через свой индивидуальный опыт, связанный с повреждениями, перенесёнными ранее. Биологи считают, что причина боли находится в повреждённых тканях. В соответствии с этим боль это ощущение, которое ассоциируется с наступившим или вероятным повреждением тканей и возникающее в какой-либо части или частях тела. Но в то же время боль всегда неприятна и поэтому представляет собой эмоциональное восприятие. Ощущения, которые имеют сходство с болью, но не являются неприятными, например, покалывание, не могут называться болью. Непрятные, необычные ощущения (дизестезия) могут считаться болью, но не во всех случаях, поскольку они могут не обладать в полной мере чувствительными качествами боли.
Многие люди отмечают боль при отсутствии тканевого поражения или других патофизиологических изменений. Обычно это происходит вследствие каких-либо психологических причин. Часто по субъективной оценке невозможно отличить их ощущение от ощущения, обусловленного повреждением тканей. Если они согласны оценивать своё ощущение как боль и, если они описывают его таким же образом, как и боль, вызванную повреждением тканей, то это ощущение должно быть признано болью. Это определение избегает связывать боль только с раздражением. Боль это не только возбуждение, вызываемое в ноцицепторах и ноцицептивных путях повреждающими стимулами, она всегда представляет собой психологическое состояние, даже, несмотря на то, что в большинстве случаев имеет непосредственную физическую причину.
Боль является симптомом при низкой интенсивности и непродолжительности (без значимого вегетативного компонента), синдром - при высокой интенсивности и продолжительности (со значимым вегетативным компонентом) и патологическим процессом - при чрезмерной интенсивности, вызывающий повреждения (болевой шок).
Процесс восприятия боли обеспечивается алгической системой или системой формирования боли («algos» в пер. с греч. «боль»).
В настоящее время более распространены термины:
Ноцицептивная система («cepere» – восприятие, «nocere» – повреждение) – система формирования боли, ее восприятия.
Антиноцицептивная система – противоболевая система.
Классификация боли
По значению:
Физиологическая - боль, имеющая адаптивное значение, сформирована в филогенезе для того, чтобы уцелеть. Двигательные реакции, боль – как стрессор, включающий, например, симпато-адреналовую реакцию (АД, глюкоза и т.д.).
Патологическая – дезадаптивное значение, имеет характер типового патологического процесса, часто даже приобретает характер самостоятельной болезни, заслоняет основное заболевание. Боль часто сопровождает воспаление.
По характеру:
Хроническая (практически всегда патологическая)
Острая (может быть и физиологической и патологической)
Первичная (локализованная) – первичная альтерация при воспалении (лезвие, игла)
Вторичная (нелокализованная) боль может быть вызвана медиаторами воспаления, повреждениями самого нерва (чувствительного, когда он сам может генерировать потенциал), возникновением псевдосинапсов между нервами и деафферентацией.
По локализации:
Местная – афферентация идет непосредственно с места повреждения, и боль совпадает с местом поражения (рецептора, нервного ствола или корешка).
Проекционная боль отмечается далеко от локализации патологического процесса, обычно она распространяется в зону иннервации, например стреляющая боль в ногах при спинной сухотке, фантомная боль (в удаленной конечности).
Иррадиирующая (или рефлекторная) боль возникает в результате передачи импульса с одной ветви нерва на другую (или в центральной нервной системе и ганглиях с одного нейрона на другой) → в результате чего в зоне иннервации последней ощущается боль. Так, например, формируются висцеро-кутанные боли: при стенокардии боль отдает в левое плечо, печеночная колика – правое плечо, почечная колика – мошонка, бедро, панкреатит вызывает опоясывающие боли, боль из толстой кишки иррадиирует в гипогастрий и нижние отделы живота. Проекция внутренних органов на поверхность тела (кожи) описывается зонами Захарьина-Геда (висцеро-сенсорные рефлексы).
По происхождению:
Ноцигенная (соматическая) боль возникает при раздражении кожных ноцицепторов, ноцицепторов глубоких тканей или внутренних органов тела, возникающие при этом импульсы, следуя по классическим анатомическим путям, достигают высших отделов нервной системы и отображаются сознанием, формируется ощущение боли.
Боль от внутренних органов возникает вследствие быстрого сокращения, спазма или растяжения гладких мышц, поскольку сами гладкие мышцы нечувствительны к жару, холоду или рассечению. Боль от внутренних органов, особенно имеющих симпатическую иннервацию, может ощущаться в определённых зонах на поверхности тела. Такая боль называется отражённой. Наиболее известные примеры отражённой боли - боль в правом плече и правой стороне шеи при поражении желчного пузыря, боль в нижней части спины при заболевании мочевого пузыря и, наконец, боль в левой руке и левой половине грудной клетки при заболеваниях сердца. Возможное объяснение состоит в том, что сегментарная иннервация внутренних органов та же, что и отдалённых областей поверхности тела. Ноцигенный тип боли терапевтически чувствителен к морфину и другим наркотическим анальгетикам и может контролироваться состоянием “ворот”.
Нейрогенная боль возникает вследствие повреждения периферической или центральной нервной системы и не объясняется раздражением ноцицепторов (ощущение боли возникает даже в здоровом органе).
Обычно отмечаются болевые ощущения в ответ на низко интенсивные, в нормальных условиях не вызывающие боли, раздражители. Например, лёгкое прикосновение, дуновение воздуха или причёсывание при тригеминальной невралгии вызывает в ответ “болевой залп”. Нейрогенная боль невосприимчива к морфину и другим опиатам в обычных анальгетических дозах.
Многие боли клинически проявляются смешанными - ноцигенными и нейрогенными элементами. Например, опухоли вызывают повреждение тканей и компрессию нервов; при диабете ноцигенная боль возникает вследствие поражения периферических сосудов, нейрогенная - вследствие нейропатии; при грыжах межпозвонкового диска,компримирующих нервный корешок, болевой синдром включает жгучий и стреляющий нейрогенный элемент.
Психогенная боль. Утверждение что боль может быть исключительно психогенного происхождения, является дискуссионным. Широко известно, что личность пациента формирует болевое ощущение. Оно усилено у истерических личностей, и более точно отражает реальность у пациентов неистероидного типа.
Люди различных этнических групп отличаются по восприятию послеоперационной боли. Пациенты европейского происхождения отмечают менее интенсивную боль, чем американские негры или латиноамериканцы. У них также отмечается низкая интенсивность боли по сравнению с азиатами, хотя эти отличия не очень значительны.
По качеству: стреляющие, рвущие, пульсирующие, сжимающие, режущие, колющие и др. (в зависимости от фантазии пациента).
Типы болевых рецепторов. Медиаторы боли. Проводниковый и центральный аппараты боли. Вегетативные и эмоциональные компоненты.
Болевые раздражения могут возникать в коже, глубоких тканях и внутренних органах. Эти раздражения воспринимаются ноцицепторами, расположенными по всему телу, за исключением головного мозга.
Болевые рецепторы
В зависимости от глубины залегания:
Поверхностные (обычно более чувствительные, и это логично, учитывая роль боли в адаптации).
Глубокие (интерорецепторы)
В зависимости от способа возбуждения:
Мономодальные реагируют на механическое воздействие.
Бимодальные реагируют на механо - и термовоздействия (менее 10, более 40).
Полимодальные реагируют на механо-, термо- и хемовоздействия.
Афферентные волокна
А-дельта волокна - миелинизированные, быстропроводящие (проводят раздражение со скоростью 6-30 м/с). Эти волокна возбуждаются высокоинтенсивными механическими (булавочный укол) и, иногда, термическими раздражениями кожи (т.е. в основном – от мономодальных рецепторов). Имеют скорее информационное значение для организма (отдернуть руку, отпрыгнуть). Анатомически А-дельта ноцицепторы представлены свободными нервными окончаниями, разветвлёнными в виде дерева (миелиновые волокна). Они располагаются, преимущественно, в коже, включая оба конца пищеварительного тракта. Находятся они также и в суставах. Трансмиттер А-дельта волокон остаётся неизвестным.
С-волокна – немиелинизированные, проводят мощные, но медленные потоки импульсации (со скоростью 0,5-2 м/с). Эти афферентные волокна представлены плотными некапсулированными гломерулярными тельцами. Они являются полимодальными ноцицепторами, поэтому реагируют как на механические, так на температурные и химические раздражения. Они активируются химическими веществами, возникающими при повреждении тканей, являясь одновременно и хеморецепторами, и считаются оптимальными тканеповреждающими рецепторами. Считается, что они предназначены для восприятия вторичной острой и хронической боли. С-волокна распределяются по всем тканям за исключением центральной нервной системы. Однако, они присутствуют в периферических нервах, как nervi nervorum. Волокна, имеющие рецепторы, воспринимающие повреждения тканей, содержат субстанцию Р, выступающую в качестве трансмиттера. Такой тип ноцицепторов также содержит calcitonin ген - связанный пептид, а волокна из внутренних органов - вазоактивный интестинальный пептид.
Первичную боль от укола, ожога (терморецепторы) – проводят А-дельта волокна, с поверхностных рецепторов, вызывают быструю, «фазическую» реакцию мышц, отдергивание. Морфин и др. наркотические аналгетики не снимает первичную боль, поэтому, например, бесполезно удалять зуб под морфиновым обезболиванием.
Вторичная боль вызывается повреждениями ткани хронического характера, чаще с хеморецепторов (под действием медиаторов), механорецепторов; из глубоких или поверхностных рецепторов, импульсы идут по С-волокнам, реакция мышц – спастическая, т.е. тоническое сокращение мышц (больной, например, «нянчит» больную руку). Морфин эффективен.
Проводящие пути
Тело I нейрона проводящего пути болевой чувствительности расположено в спино-мозговых узлах, II нейрона – в задних рогах спинного мозга, III нейрона – в таламусе. Далее импульсы могут расходиться веером – в корковый анализатор, вегетативные подкорковые центры.
Проведения зубной боли имеет некоторые особенности. Так основная афферентная импульсация идет по тройничному нерву, поэтому тело I нейрона находится в тройничном (тригеминальном) ганглии, а не в спино-мозговом. Тело II нейрона расположено в чувствительные ядрах тройничного нерва.
Второй по значимости – языкоглоточный нерв (задняя часть языка, глотки, миндалин, мягкое небо). Третий – промежуточная ветвь лицевого нерва, он имеет свой ганглий, путь проходит через ядро единичного пути.
Но для всех нервов – переключение в (тело III нейрона).
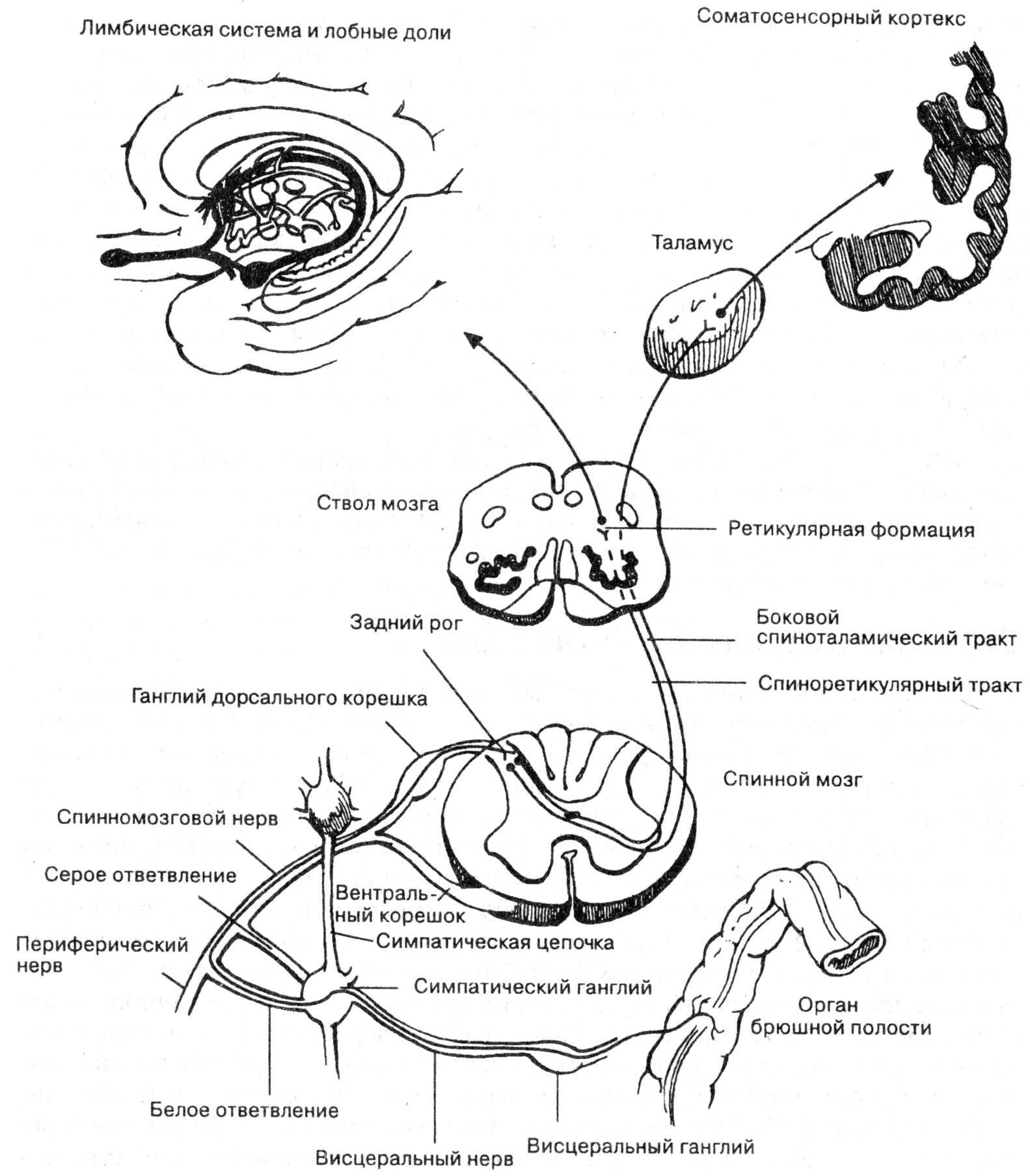
Рис. 8.1. Трехнейронный проводящий путь болевой чувствительности.
Спино-таламические
Специфический (неоспиноталамический) путь - задние рога спинного мозга специфические ядра таламуса кора задней центральной извилины. Этот путь является малонейронным, быстрым, проводит по А-дельта волокнам пороговую, эмоционально неокрашенную, точно локализованную боль (эпикритическая боль).
Неспецифический (палеоспиноталамический) путь - задние рога спинного мозга неспецифические ядра таламуса кора лобной и теменной долей диффузно. Проводит подпороговую, эмоционально окрашенную, плохо локализованную боль (протопатическая боль). Является медленным (по С-волокнам), многонейронным, т. к. образует многочисленные коллатерали к продолговатому мозгу, ретикулярной формации, лимбической системе, гиппокампу. Подпороговые болевые импульсы подвергаются суммации в таламусе.
Тригемино-таламический
Вегетативные и эмоциональные компоненты боли
Проводимые по неспецифическому болевому пути импульсы возбуждают эмоциональные центры лимбической системы (эмоции в подавляющем большинстве случаев отрицательные), вегетативные центры гипоталамуса, продолговатого мозга. Поэтому боль сопровождается страхом, тягостными переживаниями, учащением дыхания, пульса, подъемом АД, расширением зрачка, саливацией, диспепсическими расстройствами.
Медиаторы ноцицептивной системы
С помощью медиаторов ноцицептивной системы информация передается с клетки на клетку.
Субстанция Р (от англ. pain – «боль») – главная.
Нейротензин.
Брадикинин.
Холецистокинин.
Глютамат.
22. – Теории возникновения боли. Механизм возникновения боли согласно теории воротного контроля. Механизмы функционирования антиноцептивной системы.
Теории возникновения боли.
Теория специфичности утверждает, что боль представляет собой отдельную сенсорную систему, в которой любой повреждающий стимул активирует специальные болевые рецепторы (ноцицепторы), передающие болевой импульс по специальным нервным путям в спинной мозг и в болевые центры головного мозга, вызывая ответную защитную реакцию, направленную на удаление от раздражителя.
Основой для создания в теории специфичности послужило учение французского философа и физиолога Р. Декарта о рефлексе. В 20-ом столетии правомерность концепции боли, как специфической проекционной сенсорной системы, была подтверждена многочисленными исследованиями и открытиями в анатомии и экспериментальной физиологии. Были обнаружены болепроводящие нервные волокна и болепроводящие пути в спинном мозге, болевые центры в различных отделах головного мозга, медиаторы боли (брадикинин, субстанция Р, ВИП и др.).
Согласно теории специфичности, психологическое ощущение боли, её восприятие и переживание признаются адекватными и пропорциональными физической травме и периферическому повреждению. В практической медицинской деятельности это положение привело к тому, что пациенты, страдающие болью и не имеющие очевидных признаков органической патологии, стали считаться "ипохондриками", "невротиками" и, в лучшем случае, направлялись на лечение к психиатру или психотерапевту.
Теория интенсивности утверждает, что ощущение боли возникает при раздражении любого рецептора избыточным стимулом (шум, свет).
Теория воротного контроля (Melzack, Wall, 1965). Поток болевой импульсации с периферии идет в задний рог спинного мозга по большим миелинизированным (А-дельта) и малым немиелинизированным (С-волокнам) нервным волокнам. Оба типа волокон образуют синапсы с нейронами второго порядка (Т) ("передача/проекция"). Когда Т-нейроны активированы, они поставляют ноцицептивную информацию в мозг.
Периферические нервные волокна также образуют синапсы с интернейронами желатинозной сустанции (ЖС), которые при стимуляции угнетают Т-нейроны. А-дельта волокна стимулируют, а С-волокна угнетают интернейроны ЖС, соответственно снижая и повышая центральную передачу ноцицептивных входящих сигналов.
Кроме того, стимуляция интернейронов ЖС на подавление активности Т-нейронов происходит через нисходящие пути, начинающиеся в центральной нервной системе (это происходит при активации различными факторами). Баланс между возбуждающими и угнетающими сигналами определяет степень передачи ноцицептивной информации в головной мозг («+» - возбуждающий сигнал; «-» - угнетающий сигнал).
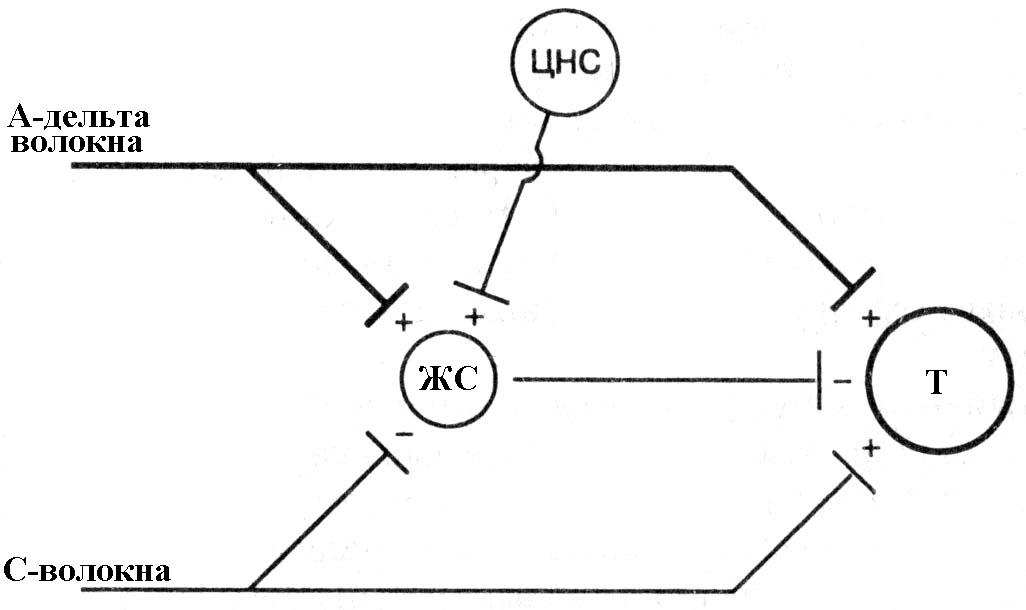
Рис. 8.2. Схема теории «воротного контроля» по R. Melzack, 1999 (объяснение в тексте).
Примечание. ЖС - желатинозная субстанция задних рогов спинного мозга, Т - трансмиссивные нейроны.
Основное научно-медицинское значение теории "входных ворот" заключалось в признании спинного и головного мозга активной системой, фильтрующей, отбирающей и воздействующей на входные сенсорные сигналы. Теория утвердила центральную нервную систему ведущим звеном в болевых процессах.
Теория «генератора патологически усиленного возбуждения» в центральной нервной системе подчеркивает значение центральных механизмов в патогенезе боли и определяет роль периферических факторов.
Генератор патологически усиленного возбуждения (ГПУВ, генератор) - это агрегат гиперактивных нейронов, продуцирующий чрезмерный неконтролируемый поток импульсов.
ГПУВ образуется в поврежденной нервной системе из первично и вторично измененных нейронов и представляет собой новую, необычную для деятельности нормальной нервной системы патологическую интеграцию, возникающую на уровне межнейрональных отношений. Особенностью генератора является его способность развивать самоподдерживающуюся активность. ГПУВ может образовываться практически во всех отделах ЦНС, его формирование и деятельность относятся к типовым патологическим процессам.
При создании генератора в системе болевой чувствительности появляются различные болевые синдромы: болевой синдром спинального происхождения (генератор в дорсальных рогах спинного мозга), тригеминальная невралгия (генератор в каудальном ядре тройничного нерва), таламический болевой синдром (генератор в ядрах таламуса).
Невромы, повреждения нервов, смещения межпозвонковых дисков вызывают боль и приводят к возникновению патологических центральных процессов. В ЦНС формируется "генератор патологически усиленного возбуждения", в результате значение периферических факторов уменьшается. Поэтому при сильной фантомной невралгической и поясничной боли после удаления невром нервов, грыж диска и т.п. устранение периферических факторов может не привести к прекращению боли.
Возникновение генератора начинается либо с первичной гиперактивации нейронов, либо с первичного нарушения их торможения. При первичной гиперактивации нейронов тормозные механизмы сохранены, но они функционально недостаточны. В этом случае имеет место вторичная недостаточность торможения, которая возрастает по мере развития генератора, при преобладании возбуждения. При первичной недостаточности тормозных механизмов появляются растормаживание и вторичная гиперактивация нейронов.
Первичная гиперактивация нейронов возникает вследствие усиленных и длительных возбуждающих воздействий: при синаптической стимуляции, при действии возбуждающих аминокислот, К+ и др. Роль синаптической стимуляции отчетливо видна на примере формирования генератора в ноцицептивной системе. Хронически раздражаемые рецепторы в тканях, эктопические очаги в поврежденных нервах, неврома (хаотически разросшиеся афферентные волокна) являются источником постоянной импульсации. Под влиянием этой импульсации в центральном аппарате ноцицептивной системы формируется генератор.
Первичное нарушение торможения нейронов формируется в условиях действия веществ, избирательно повреждающих тормозные процессы. Такой эффект имеет место при действии столбнячного токсина, нарушающего выделение пресинаптическими окончаниями тормозных медиаторов; при действии стрихнина, блокирующего глициновые рецепторы на постсинаптических нейронах спинного мозга, где глицин оказывает тормозной эффект; при действии некоторых конвульсантов, нарушающих постсинаптическое торможение.
Поскольку деятельность генераторных механизмов определяется множественными взаимодействиями, влиять на нее можно путем одновременного использования антидепрессантов, раздражения триггерных точек электрическим током, физиотерапией и др.
Понятие антиноцицептивной системы. Ее уровни, медиаторы.
Антиноцицептивная система
Комплекс ноцицептивной системы в равной степени сбалансирован в организме комплексом антиноцицептивной системы, обеспечивающей контроль за активностью структур, участвующих в восприятии, проведении и анализе болевых сигналов.
В настоящее время установлено, что болевые сигналы, поступающие с периферии, стимулируют активность различных отделов центральной нервной системы (околопроводное серое вещество, ядра шва ствола мозга, ядра ретикулярной формации, ядра таламуса, внутренней капсулы, мозжечка, интернейроны задних рогов спинного мозга и др.), оказывающих нисходящее тормозное действие на передачу ноцицептивной афферентации в дорзальных рогах спинного мозга.
Основные нейроны антиноцецептивной системы локализованы в околоводопроводном сером веществе (сильвиев водопровод соединяет III и IV желудочки). Их аксоны образуют нисходящие пути к продолговатому и спинному мозгу и восходящие пути к ретикулярной формации, таламусу, гипоталамусу, лимбической системе, базальным ганглиям и коре.
Медиаторами этих нейронов являются пентапептиды: метэнкефалин и лейэнкефалин. Энкефалины возбуждают опиатные рецепторы. Опиатные рецепторы возбуждаются не только медиаторами-энкефалинами, но и другими компонентами антиноцецептивной системы – гормонами головного мозга - эндорфинами (бета-эндорфин, динорфин).
В механизмах развития анальгезии наибольшее значение придаётся серотонинергической, норадренергической, ГАМКергической и опиоидергической системам мозга.
Основная из них, опиоидергическая система, образована нейронами, тело и отростки которых содержат опиоидные пептиды (бета-эндорфин, мет-энкефалин, лей-энкефалин, динорфин).
Связываясь с определёнными группами специфических опиоидных рецепторов (мю-, дельта- и каппа-опиоидные рецепторы), 90% которых расположено в дорзальных рогах спинного мозга, они способствуют высвобождению различных химических веществ (гамма-аминомасляная кислота), тормозящих передачу болевых импульсов.
Энкефалины и эндорфины возбуждают опиатные рецепторы. В энкефалинергических синапсах опиатные рецепторы находятся на постсинаптической мембране, но эта же мембрана является пресинаптической для других синапсов. Опиатные рецепторы ассоциированы с аденилатциклазой и вызывают ее ингибирование, нарушая в нейронах синтез цАМФ. В итоге уменьшается вход кальция и освобождение медиаторов, включая медиаторы боли (субстанция P, холецистокинин, соматостатин, глутаминовая кислота).
К медиаторам антиноцецептивной системы относятся также катехоламины. Они возбуждают тормозные 2-адренорецепторы, осуществляя тем самым постсинаптическое торможение боли.
Виды клеточного торможения
Пресинаптическое направленно на торможение выделения медиатора из-за гиперполяризации всего нейрона.
Постсинаптическое – гиперполяризация следующего нейрона.
Говоря об антиноцицептивной системе, первым компонентом следует ставить:
Желатинозную субстанцию спинного мозга (в чувствительных ядрах тригеминуса видимо, есть нечто подобное).
Нисходящие гипоталамо-спинальные пути (возможность обезболивания путем гипноза, внушения и самовнушения). С аксонов в спинном мозгу или на ядрах тригеминуса тоже выделяются тормозные медиаторы.
Естественная болеутоляющая система так же важна для нормальной жизнедеятельности, как и болесигнализирующая система. Благодаря ей, незначительные повреждения типа ушиба пальца или растяжения связок вызывают сильные болевые ощущения только на короткое время - от несколько минут до нескольких часов, не заставляя нас страдать в течение дней и недель, что случилось бы в условиях сохранения боли до полного заживления.
Таким образом, физиологическая ноцицепция включает четыре основных процесса:
Трансдукцию - процесс, при котором повреждающее воздействие трансформируется в виде электрической активности в свободных неинкапсулированных нервных окончаниях (ноцицепторах). Их активация происходит либо вследствие прямых механических или термических стимулов, либо под воздействием эндогенных тканевых и плазменных алгогенов, образующихся при травме или воспалении (гистамин, серотонин, простагландины, простациклины, цитокины, ионы К+ и Н+ , брадикинин).
Трансмиссию - проведение возникших импульсов по системе чувствительных нервных волокон и путей в центральную нервную систему (тонкие миелиновые А-дельта и тонкие безмиелиновые С-афференты в составе аксонов спинномозговых ганглиев и задних спинномозговых корешков, спиноталамические, спиномезенцефалические и спиноретикулярные пути, идущие от нейронов задних рогов спинного мозга к образованиям таламуса и лимбико-ретикулярного комплекса, таламокортикальные пути к соматосенсорным и фронтальной зонам коры головного мозга).
Модуляцию - процесс изменения ноцицептивной информации нисходящими, антиноцицептивными влияниями центральной нервной системы, мишенью которых являются преимущественно нейроны задних рогов спинного мозга (опиоидергические и моноаминовые нейрохимические антиноцицептивные системы и система воротного контроля).
Перцепцию - субъективное эмоциональное ощущение, воспринимаемое как боль и формирующееся под воздействием фоновых генетически детерминированных свойств центральной нервной системы и ситуационно меняющихся раздражений с периферии.
23. - Экстремальные состояния. Отличия обморака, колапса, шока и комы. общий патогенез шока.
Экстремальные состояния - состояния, сопровождающиеся грубыми расстройствами метаболизма и жизненно важных функций и представляющие непосредственную опасность для жизни.
Экстремальные состояния часто связаны с действием сверхсильных патогенных факторов.
Виды экстремальных состояний
коллапс,
обморок,
шок,
кома,
терминальные состояния (иногда включаются).
Шок – стадийно протекающее острое состояние, возникающее вследствие воздействия сверхсильных стрессоров и характеризующееся гиповолемией, снижением сердечного выброса и АД, нарушениями микроциркуляции и гипоксией.
Стадии шока
Эректильная (напряжения) – сопровождается эмоциональным и поведенческирм возбуждением, активацией органов и систем.
Торпидная – угнетение психической, эмоциональной и поведенческой деятельности.
Терминальная.
Этиологическим фактором шока является любой сверхсильный раздражитель.
Общий патогенез шока
Первичное звено – активация симапато-адреналовой и гипоталамо-гипофизарно-надпочечниковой систем, что обуславливает симптомы эректильной фазы (поведение, увеличение силы и частоты сердечных сокращений, дыхания, усиление энергообмена, активация глюконеогенеза).
Торможение (угнетение) ЦНС на начальном этапе охранительное, но при действии очень сильных раздражителей переходит в патологическое (запредельное), обуславливает торпидную фазу.
«Централизация кровообращения» - циркуляция крови по системе "сердце-мозг-сердце". Необходимо для сохранения кровотока в жизненно важных органах и поддержания системного АД. Развивается в результате активации симапато-адреналовой системы (вазоконстрикция сосудов -органов [кожа, почки, органы брюшной полости], вазодилатация сосудов -органов [сердце, мозг]).
Основное звено патогенеза - гиповолемия (и падение АД) вследствие снижения сердечного выброса и недостаточной вазоконстрикции (приводит к нарушению микроциркуляции).
Гиповолемия – несоответствие между объемом сосудистого русла и объемом циркулирующей крови (проявлением ее чаще всего бывает ↓АД).
Вазоконстрикция сосудов -органов приводит к ишемическому стазу, вследствие чего развивается гипоксия, ацидоз. Образующиеся при этом вазоактивные соединения вызывают неадекватную вазодилятацию, в результате которой формируется стаз, сладж, ДВС-синдром, лежащие в основе «шоковых органов» (почки, легкие, печень).
- полное выключение сознания с грубыми расстройствами рефлекторной сферы, вплоть до арефлексии и отсутствия болевой чувствительности. Коме может предшествовать ступор и сопор.
Понятия обморока и коллапса. Отличия от шока.
Коллапс – остро возникающая сосудистая недостаточность, обусловленная первичным дефицитом вазоконстрикции, сопровождающаяся гиповолемией и падением АД.
Коллапс сопровождается расстройством сознания (например, «мушки» перед глазами). Потеря сознания при коллапсе свидетельствует о его переходе в обморок.
Обморок (синкопе, синкопальное состояние) – внезапная кратковременная потеря сознания, обусловленная недостаточностью церебрального биоокисления и гипоксией мозга.
Ступор (оглушение) – расстройство сознания с сохранением словесного контакта.
Сопор – выключение сознания с отсутствием словесного контакта и сохранением реакции на боль.
24. – Особенности патогенеза шоков различной этиологии.
Особенности шоков в зависимости от их этиологии
Травматический шок – представляет собой в большинстве случаев комбинацию болевого и геморрагического шоков.
Болевой шок – сильный болевой раздражитель запредельное торможение ЦНС распространение торможения на сосудодвигательный центр головного мозга нарушение регуляции сосудистого тонуса падениеАД.
Геморрагический шок – гиповолемия обусловлена массивной кровопотерей.
Ожоговый шок [возникает при поражении более 15% площади поверхности тела ожогами II степени и более] – гиповолемия возникает из-за
значительных потерей плазмы через ожоговую поверхность;
интоксикации [продукты распада поврежденных тканей, токсины микроорганизмов при присоединении инфекции];
грубых изменений физико-химических свойств крови гемолиз эритроцитов.
Анафилактический шок – обусловлен нарушением иммунологической реактивности и развитием аллергической реакции I типа; взаимодействие аллергена с Ig E, фиксированными на поверхности тучных клеток высвобождение гистамина бронхоспазм [острая дыхательная недостаточность] + повышение проницаемости сосудов отек легких и выход жидкости за пределы сосудистого русла (гиповолемия). Смерть при анафилаксии наступает быстро именно из-за дыхательной недостаточности, выраженная гиповолемия просто не успевает развиться.
Следует сказать о возможности возникновения лекарственного, ятрогенного шока. Многие, даже небелковые, низкомолекулярные препараты (гаптены) могут после соединения с белками организма приобретать свойства полных антигенов и при повторном введении вызывать массивную аллергическую реакцию, сопровождающуюся значительным выходом гистамина, оказывающего выраженное сосудорасширяющее действие.
Гемотрансфузионный шок – возникает при переливании несовместимой крови массивная агглютинация и гемолиз эритроцитов гемическая гипоксия + высвобождение эритропластина [обладает активностью тромбопластина, хотя и меньшей] развитие ДВС-синдрома. Важно развитие гемоглобинурийного нефроза – закупорка канальцев почки кислым хромопротеидом почечная недостаточность.
Кардиогенный шок – возникает при ИМ, аритмиях, тампонаде сердца, ТЭЛА. Различают виды:
Болевой шок.
Истиный шок – обусловлен снижением насосной функции сердца из-за повреждения 50-65% миокарда.
Аритмический шок - обусловлен снижением насосной функции сердца из-за некоординированных сердечных сокращений.
Септический (эндотоксический) шок – связан с массивным выбросом фагоцитами различных интерлейкинов [в первую очередь фактора некроза опухоли (ФНО)] дилатация сосудов + повышение сосудистой проницаемости выход жидкости за пределы сосудистого русла. Кроме того, эндотоксины возбудителей активируют протеолитические системы (калликреин-кининовую, фибринолитическую) ДВС-синдром.
Турникетный шок [возникает при сдавлении мягких тканей более 4 часов], сопровождает «краш-синдром», обусловлен болевым синдромом и интоксикацией продуктами распада после длительной ишемии. Кроме того, миоглобин вызывает закупорку канальцев почки почечная недостаточность.
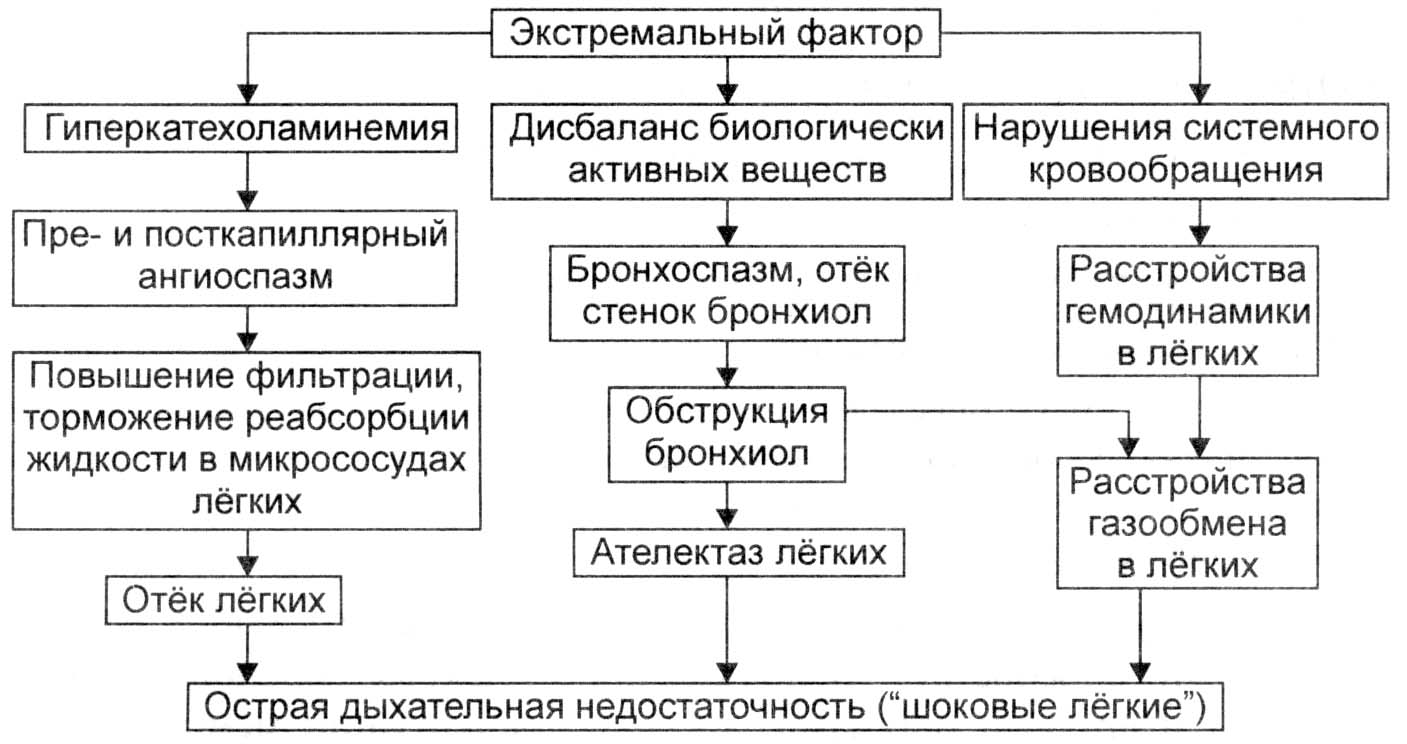
Рис. 9.1. Патогенез развития синдрома «шоковое легкое».
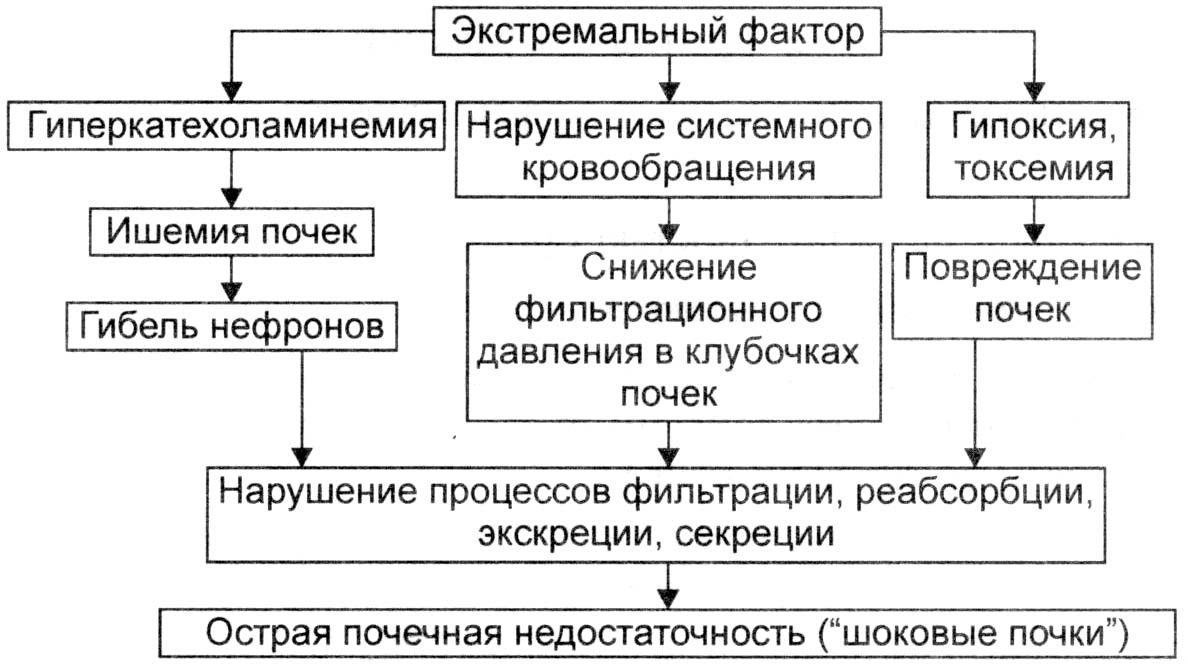
Рис. 9.1. Патогенез развития синдрома «шоковая почка».
25. – Воспаление. Этиология. Виды и стадии. Основные звенья патогенеза воспаления. Проявления воспаления. Принципы терапии.
Воспаление - типовой патологический процесс, защитно-приспособительная реакция, развивающаяся в ответ на действие флогогенного агента, направленная на устранение и локализацию этого агента, и восстановление ткани, хотя может привести к их повреждению.
Воспалительные заболевания составляют около 80% всей патологии в практике врача любой специальности, дают наибольшее число дней нетрудоспособности.
Классификация воспаления По этиологии воспаления (в зависимости от вида флогогенного агента):
Экзогенные факторы:
Механические.
Физические (лучевая, электрическая энергия, тепло, холод).
Химические (кислоты, щелочи).
Биологические (бактерии, вирусы, риккетсии, паразиты, простейшие).
Антигенные (аллергическое воспаление).
Эндогенные факторы:
Продукты тканевого распада - инфаркт, некроз, кровоизлияние.
Тромбоз и эмболия.
Продукты нарушенного метаболизма - токсические или биологически активные вещества (например, при уремии токсические вещества, образующиеся в организме, выделяются из крови слизистыми оболочками, кожей, почками и вызывают в этих тканях воспалительную реакцию).
Отложение солей или выпадение биологических соединений в виде кристаллов.
Нервно-дистрофические процессы.
По участию микроорганизмов:
Инфекционное (септическое).
Неинфекционное (асептическое).
По реактивности:
Гиперэргическое.
Нормэргическое.
Гипоэргическое.
По течению:
Острое.
Подострое.
Хроническое.
По преобладанию стадии:
Альтеративное возникает в паренхиматозных органах (в последнее время отрицается).
Экссудативное возникает в клетчатке и сосудах (крупозное, серозное, фибринозное, гнойное, гнилостное, геморрагическое, катаральное, смешанное).
Пролиферативное (продуктивное) возникает в костной ткани.
Стадии воспаления
Стадия альтерации (повреждение) бывает:
первичная,
вторичная.
Стадия экссудации в неё входят:
сосудистые реакции,
собственно экссудация,
маргинация и эмиграция лейкоцитов,
внесосудистые реакции (хемотаксис и фагоцитоз).
Стадия пролиферации (восстановление поврежденных тканей):
Аутохтонность - это свойство воспаления раз начавшись, протекать через все стадии до логического завершения, т.е. включается каскадный механизм, когда предыдущая стадия порождает последующую.
Местные признаки воспаления были описаны римским энциклопедистом Цельсом. Он назвал 4 признака воспаления: краснота (rubor), припухлость (tumor), местный жар (color), боль (dolor). Пятый признак назвал Гален - это нарушение функции - functio laesa.
Покраснение связано с развитием артериальной гиперемии и "артериализацией" венозной крови в очаге воспаления.
Жар обусловлен увеличенным притоком теплой крови, активацией метаболизма, разобщением процессов биологического окисления.
"Опухоль" ("припухлость") возникает вследствие развития экссудации и отека, набухания тканевых элементов, увеличения суммарного диаметра сосудистого русла в очаге воспаления.
Боль развивается в результате раздражения нервных окончаний различными биологически активными веществами (гистамин, серотонин, брадикинин и др.), сдвига активной реакции среды в кислую сторону, возникновения дисионии, повышения осмотического давления и механического растяжения или сдавления тканей.
Нарушение функции воспаленного органа связано с расстройством его нейроэндокринной регуляции, развитием боли, структурными повреждениями.
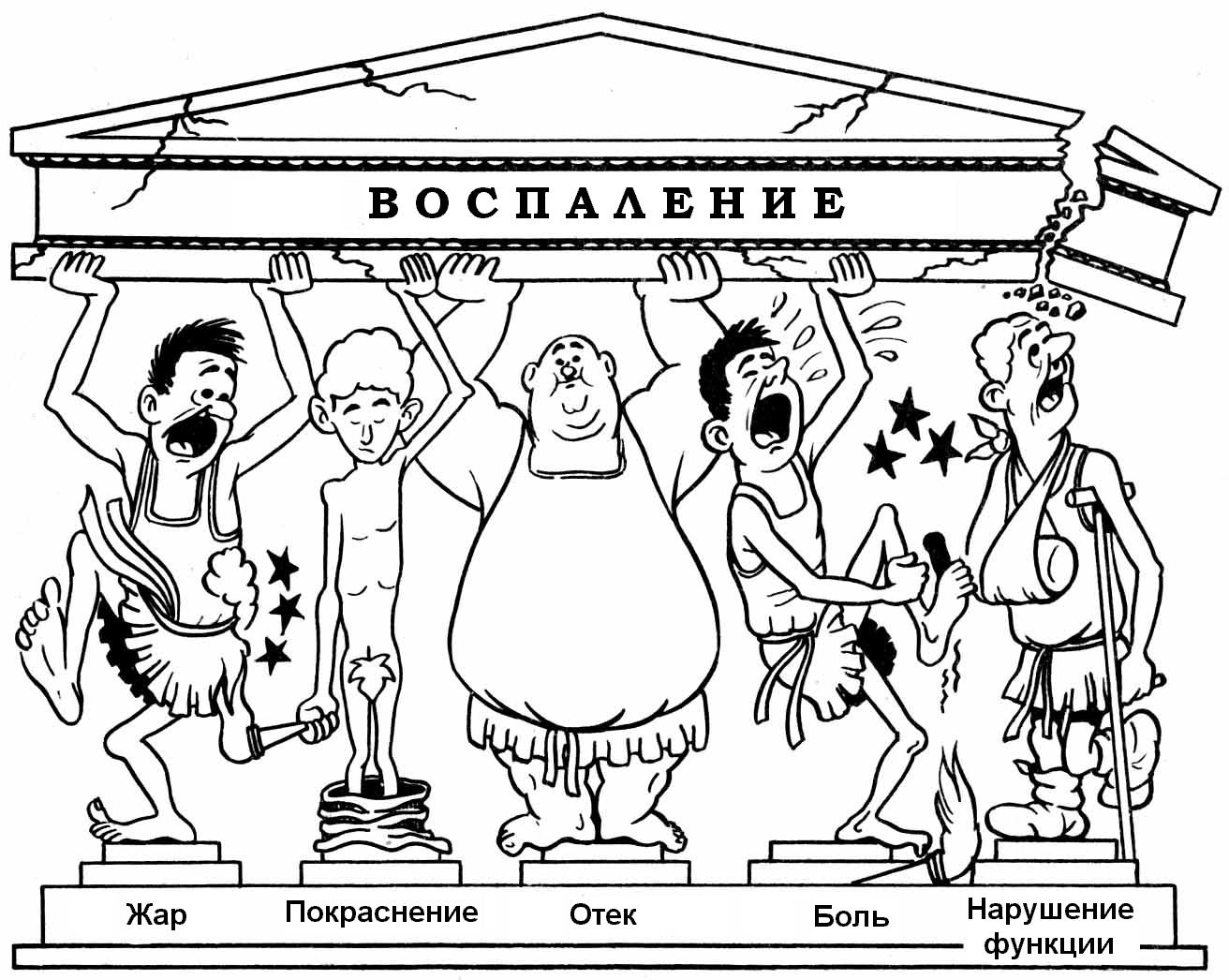
Рис. 10.1. Карикатура P. Cull на описание доктором A. A. Willoughby классических местных признаков воспаления.
Общие признаки воспаления
Изменение количества лейкоцитов в периферической крови: лейкоцитоз (развивается при подавляющем большинстве воспалительных процессов) или значительно реже лейкопения (например, при воспалении вирусного происхождения). Лейкоцитоз обусловлен активацией лейкопоэза и перераспределением лейкоцитов в кровеносном русле. К числу основных причин его развития относятся стимуляция САР, воздействие некоторых бактериальных токсинов, продуктов тканевого распада, а также ряда медиаторов воспаления (например, ИЛ1, фактора индукции моноцитопоэза и др.).
Лихорадка развивается под влиянием поступающих из очага воспаления пирогенных факторов, таких как липополисахариды, катионные белки, ИЛ1 и др.
Изменение белкового “профиля” крови выражается в том, что при остром процессе в крови накапливаются синтезируемые печенью так называемые “белки острой фазы” (БОФ) воспаления - С-реактивный белок, церулоплазмин, гаптоглобин, компоненты комплемента и др. Для хронического течения воспаления характерно увеличение в крови содержания - и особенно -глобулинов.
Изменения ферментного состава крови выражаются в увеличении активности трансаминаз (например, аланинтрансаминазы при гепатите; аспартаттрансаминазы при миокардите), гиалуронидазы, тромбокиназы и т.д.
Увеличение скорости оседания эритроцитов (СОЭ) из-за снижения отрицательного заряда эритроцитов, повышения вязкости крови, агломерации эритроцитов, изменения белкового спектра крови, подъема температуры.
Изменения содержания гормонов в крови заключаются, как правило, в увеличении концентрации катехоламинов, кортикостероидов.
Изменения в иммунной системе и аллергизация организма выражаются в нарастании титра антител, появлении сенсибилизированных лимфоцитов в крови, развитии местных и общих аллергических реакций.
Принципы противовоспалительной терапии.
Симптоматическая терапия направлена на снятие основных классических признаков воспаления
обезболивающих - для снятия боли,
мочегонных - для снятия отека,
сосудосуживающих - для уменьшения гиперемии и отека.
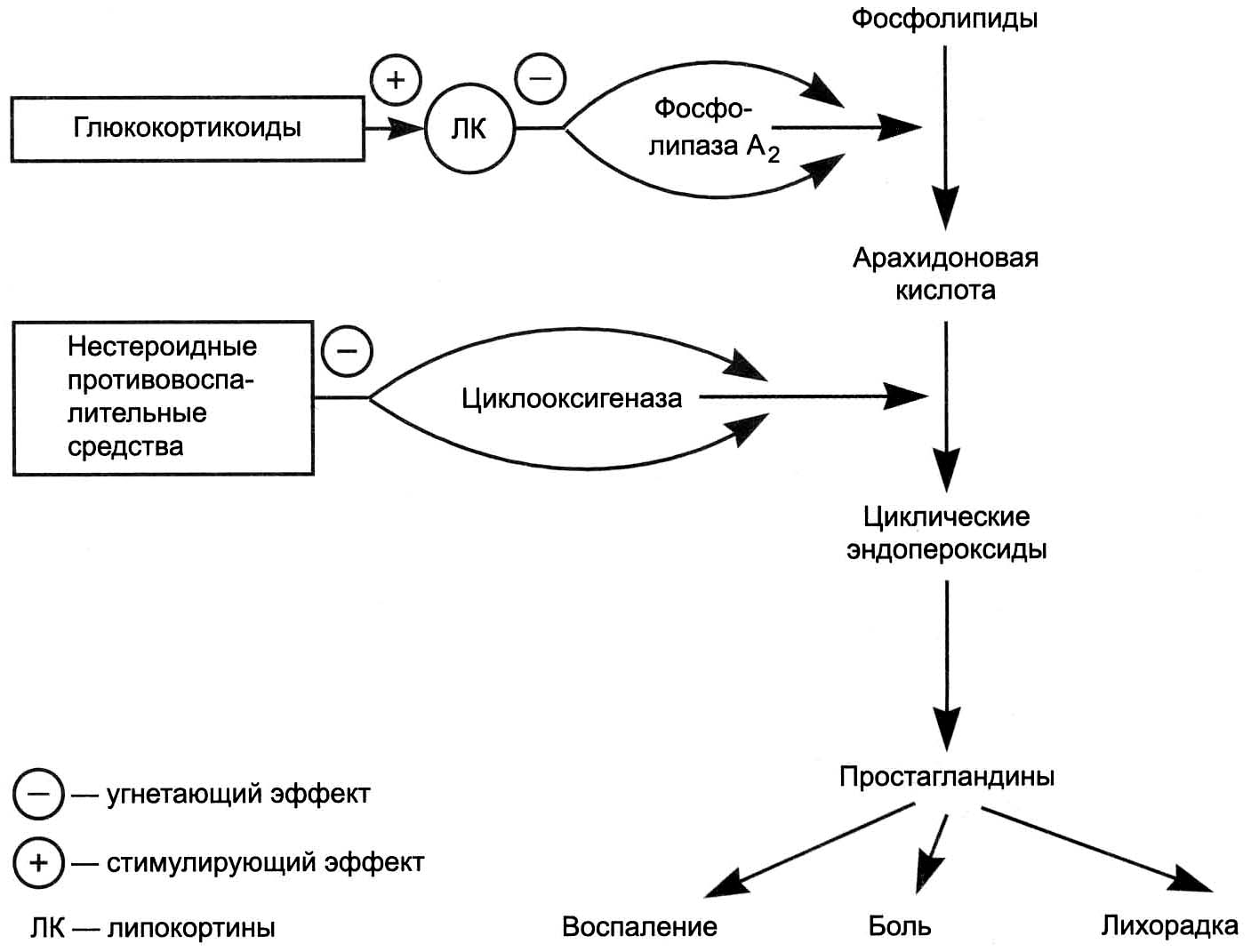
Рис. 10.8. Схема влияния противовоспалительных средств на биосинтез простагландинов.
Патогенетическая терапия включает подходы для уменьшения синтеза медиаторов воспаления (брадикинина, простагландинов, гистамина и др.) или блокаду рецепторов для этих медиаторов.
Препараты глюкокортикоидов (преднизолон, гидрокортизон), которые благодаря своему катаболическому действию тормозят синтез ферментов, необходимых прямо или косвенно для синтеза медиаторов воспаления (фосфолипаз, липоксигеназы и циклоксигеназы, гистидиндекарбоксилазы, калликреина и т.д.). Глюкокортикоиды особенно хорошо на разных ступенях тормозят иммунные и аутоиммунные воспаления, но опасны при инфекционных воспалениях.
Нестероидные противовоспалительные средства, блокирующие синтез простагландинов за счет угнетения циклооксигеназы (индометацин, вольтарен, ацетилсалициловая кислота).
ингибиторы синтеза лейкотриенов путем блокады липоксигеназы (зилеутин).
Разрабатываются препараты, блокирующие простагландиновые и лейкотриеновые рецепторы (сулотробан, зилеутин).
Лечение воспаления может включать назначение антиоксидантов - препаратов витамина Е, а также препаратов, связывающих двухвалентное железо (деферроксамин, аполактоферрин), поскольку оно участвует в образовании активных форм кислорода – важного медиатора воспаления.
Если воспаление сопровождается деструктивным процессами (абсцессы легких) оправдано назначение (особенно при введение непосредственно в зону процесса) антиферментных препаратов ингибиторов протеаз (гордокс, контрикал).
Учитывая, что воспаление не только патологический, но и адаптивный процесс, при некоторых вялотекущих (гипоэргических) воспалениях применяют приемы, активирующие данный процесс иным путем, например через усиление кровотока (местнораздражающие для включения кутано-висцеральных рефлексов), стимуляцию лейкопоэза и фагоцитоза (препараты-обломки нуклеиновых кислот и витамины), пирогенного действия (пирогенал). Фазу пролиферации можно усилить назначением препаратов, стимулирующих белковый синтез прямо (стероидные или нестероидные анаболики) или косвенно (витамины, препараты аминокислот). Но если при значительном повреждении тканей фаза пролиферации воспаления заканчивается избыточным развитием соединительной ткани, для рассасывания рубцов применяются ферментные препараты, вводимые путем инъекции или электрофореза (лидаза, ронидаза).
26. – Альтерация. Виды. Медиаторы воспаления: виды, происхождение и значение в патогенезе восполения.
Первичная альтерация вызывается непосредственным действием повреждающего агента (например, механическая травма молотком).
Для неё характерны ацидоз повреждения, снижение макроэргов, нарушение работы насосов, накопление недоокисленных продуктов, изменение рН, повышение проницаемости мембранных структур, набухание клетки.
Вторичная альтерация возникает в динамике воспалительного процесса и обусловлена как воздействием флогогенного агента, так и факторов первичной альтерации (в основном нарушениями кровообращения).
Для неё характерно непосредственное воздействие лизосомальных ферментов (гидролазы, фосфолипазы, пептидазы, коллагеназы и т.д.), их повреждающее влияние. Опосредованное действие оказывают медиаторы, система комплемента, кининовая система.
Проявления альтерации:
Нарушение биоэнергетических процессов в тканях.
Отвечают на повреждение все элементы поврежденной ткани: микроциркуляторные единицы (артериолы, капилляры, венулы), соединительная ткань (волокнистые структуры и клетки), тучные клетки, нервные клетки.
Нарушение биоэнергетики в этом комплексе проявляются в снижении потребления кислорода тканью, снижении тканевого дыхания. Повреждение митохондрий клеток является важнейшей предпосылкой для этих нарушений.
В тканях преобладает гликолиз. В результате возникает дефицит АТФ, дефицит энергии. Преобладание гликолиза ведет к накоплению недоокисленных продуктов (молочной кислоты), возникает ацидоз.
Развитие ацидоза в свою очередь приводит к нарушению активности ферментных систем, к дезорганизации метаболического процесса.
Нарушение транспортных систем в поврежденной ткани.
Это связано с повреждением мембран, недостатком АТФ, необходимой для функционирования калий-натриевого насоса.
Универсальным проявлением повреждения любой ткани всегда будет выход калия из клеток, и задержка в клетках натрия. С задержкой натрия в клетках связано еще одно тяжелое или летальное повреждение - задержка в клетках воды, то есть внутриклеточный отек.
Выход калия ведет к углублению процесса дезорганизации метаболизма, стимулирует процессы образования биологически активных веществ - медиаторов.
Повреждение мембран лизосом.
При этом высвобождаются лизосомальные ферменты. Спектр действия лизосомальных ферментов чрезвычайно широк, фактически лизосомальные ферменты могут разрушать любые органические субстраты. Поэтому при их высвобождении наблюдаются летальные повреждения клеток.
Кроме этого лизосомальные ферменты, действуя на субстраты, образуют новые биологические активные вещества, токсические действующие на клетки, усиливающие воспалительную реакцию - это лизосомные флогогенные вещества.
При альтерации возможны метаболические (гипоксия) или структурные изменения (механическая травма), поэтому выделяют два ее патогенетических механизма:
повреждение биоэнергетики (ишемия, гипоксия),
повреждение мембран и транспортных систем.
Большую роль в механизме воспаления играют медиаторы. Их делят на: провоспалительные и противовоспалительные, локальные (тканевые), циркулирующие и промежуточные. Локальные (тканевые) медиаторы
Гистамин выбрасывается при дегрануляции тучных клеток (расширение сосудов микроциркуляторного русла, повышение проницаемости микрососудов).
Серотонин может выделяться из тучных клеток, но главным его источником являются тромбоциты (медиатор боли, сосудистые эффекты меняются в зависимости от количества: в физиологических условиях является вазоконстриктором, в высоких концентрациях, при воспалении, - вазодилататором, повышает проницаемость сосудов).
Простагландины - это местные гормоны, модуляторы клеточных процессов, коротко живущий чрезвычайно химически активный класс.
простагландины класса Е (Е1, Е2) расширяют сосуды, повышают их проницаемости, являются медиаторами боли;
простагландины класса F обладают противовоспалительным эффектом;
простациклин (простагландин I2), его источником являются тромбоциты (расширяет сосуды, препятствует тромбообразованию).
тромбоксан (стимулирует тромбообразование, вызывает вазоконстрикцию, способствует агрегации клеток крови).
Лейкотриены образуются в нейтрофилах, эозинофилах, Т-лимфоцитах (стимулируют хемотаксис, особенно активен лейкотриен В4).
Циркулирующие медиаторы (образуются из неактивных белковых предшественников)
Кинины (брадикинин и калидин) образуются из кининогенов под действием ферментов калликреинов. Брадикинин и калидин способствуют расширению микрососудов, повышению проницаемости. Брадикинин является важнейшим медиатором боли.
Система комплемента - отдельные элементы этой системы по-разному влияют на развитие воспаления. Хемотаксическим эффектом обладают С3 и С5 компоненты. Кроме того, компоненты комплемента опосредовано влияют на проницаемость сосудистой стенки, и имеется их взаимосвязь с системой кининов.
Система гемостаза и особенно фактор Хагемана относятся к пусковому механизму свертывания крови. Фактор Хагемана при воспалении активирует коагуляцию, кининогенез и систему комплемента, кроме того, он регулирует активность фибринолитической системы.
Промежуточные медиаторы
Приносятся в очаг воспаления лейкоцитами. В очаг воспаления поступают нейтрофилы (микрофаги), они высвобождают лизосомальные ферменты, простогландины. Медиаторы, которые выделяют моноциты, объединены общим терминов монокины. Они высвобождают также защитные белки: интерфероны, стимуляторы иммунной системы - интерлейкины. Лимфоциты высвобождают лимфокины.
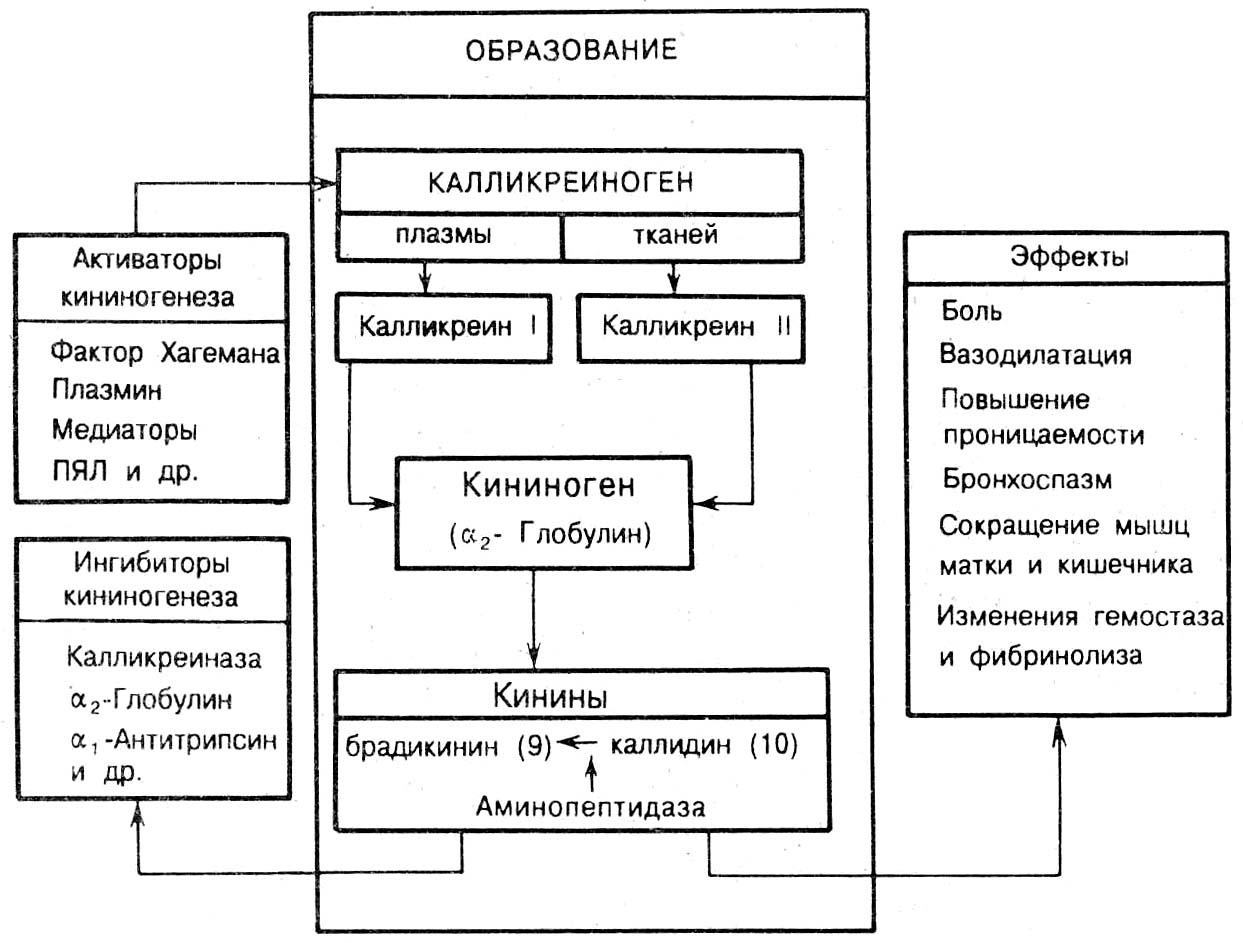
Рис. 10.2. Схема активации, ингибирования и эффектов кининовой системы в очаге воспаления (в скобках указано количество аминокислот в составе кинина).
Функции медиаторов воспаления
Боль (брадикинин, Н+, К+, отёк).
Тонус и проницаемость сосудов.
Свёртываемость крови (контактная система, тромбоксан, простациклин).
Хемотаксис.
Фагоцитоз.
Пролиферация.
Регулирование иммунной системы.
Разрешение воспаления.
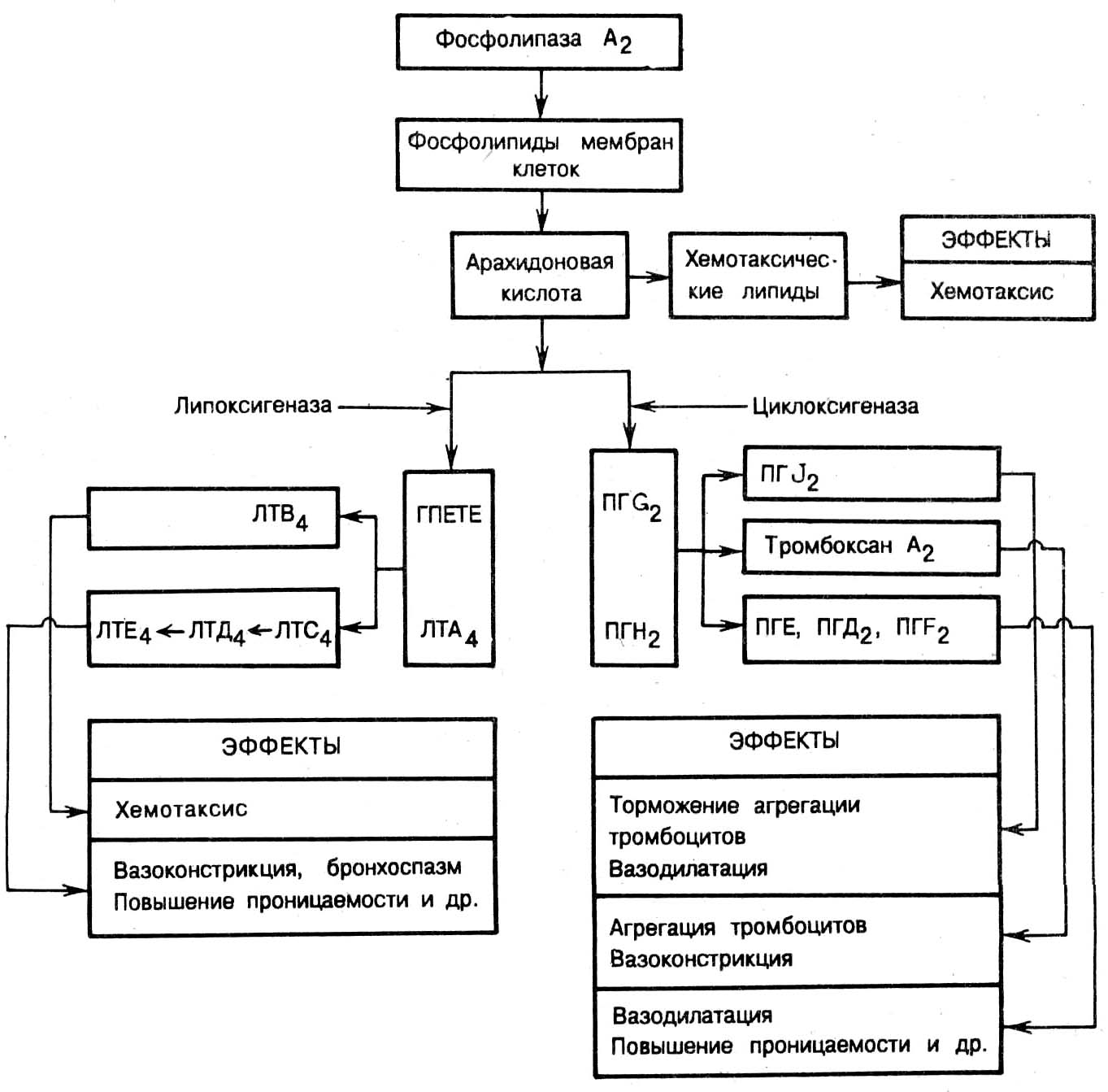
Рис. 10.3. Схема арахидонового каскада. Образование простагландинов (ПГ), лейкотриенов (ЛТ) и хемотаксических липидов, их действие в очаге воспаления. ГПЕТЕ - гидропероксид эйкозотетраеновой кислоты.
Источники медиаторов
Нервные окончания (ацетилхолин, серотонин, катехоламины).
Тучные клетки, базофилы (гистамин).
Микро- и макрофаги: «дворники», которые во время работы дают сигналы соседним клеткам и дистантно, с помощью медиаторов запускают отдельные реакции.
Лимфоциты играют важную роль в иммунном воспалении.
Тромбоцитам принадлежит особая роль в гемостазе + их трофическая роль (трофоциты) на сосуды, которая определяется тромбоцитарным фактором роста.
Эозинофилы - им принадлежит киллерная роль, это перекись-медиаторы и инактиваторы гистамина (гистаминаза) в финале воспаления. “Алая зоря выздоровления”.
Иногда медиаторы называют собирательно (в зависимости от клетки-источника): лейкокины, монокины. Многие медиаторы такого типа могут действовать и местно и дистантно, “зовут” другие клетки в очаг, усиливают их синтез.
Классификация медиаторов по химической структуре
Биогенные амины – самые ранние медиаторы (гистамин, адреналин, серотонин, полиамины - спермин, путресцин).
Гистамин образуется в тучных клетках из гистидина под влиянием гистидин-декарбоксилазы и находится в гранулах.
Серотонин (5-гидрокситриптамин) образуется при декарбоксилировании триптофана, в основном в клетках энтерохромафинной системы (ЖКТ, мозг), выделяется в основном из тромбоцитов.
Катехоламины играют значительную роль при стрессе.
Полиамины (путресцин, спермин) стимулируют пролиферацию.
Пептиды (олигопептиды и белки)
Калликренин-кининовая система.
Лизосомальные ферменты.
Система комплемента - система сывороточных белков, обозначающихся С1-С9, имеющих каскадный принцип активации. При этом наблюдается повреждение мембран, дегрануляция, хемотаксис, маргинация, опсонизация. Альтернативный путь можно увидеть при рассмотрении С3, С5.
Свертывающая и фибринолитическая системы.
Лейкокинины - пептиды некининового происхождения, образующиеся из фрагментов иммуноглобулинов под влиянием нейтрофильной и макрофагальной лейко-кининогеназы. Тафтсин стимулирует фагоцитоз, пролиферацию Т-лимфоцитов
Лимфо- и монокины. Интерлейкины играют важную роль в пирогении, оказывают стимуляцию пролиферации. Интерфероны имеют противовирусную активность, стимулируют фагоцитоз.
Лизоцим обладает бактерицидным эффектом.
Липидные медиаторы.
Простагландины - их делят на констрикторы (действуют через кальцевые каналы) и дилятаторы (действуют через систему аденилатциклазы) сосудов, они по-разному влияют на гемостаз (агрегация тромбоцитов). Тромбоксан (А2), простациклин (I2), простагландин Е.
Лейкотриены – участвуют в хемотаксисе, увеличении проницаемости мембран.
Фактор активации тромбоцитов - способствует маргинации.
Другие группы: гепарин, перекиси, свободные радикалы (оксид азота, супероксидный радикал).
Основные медиаторы альтерации
Система комплемента.
Свободные радикалы.
Лизосомальные ферменты.
27. – Экссудация. Сосудистые реакции. Механизмы экстравазации жидкости и клеток.
Основные медиаторы экссудации
Гистамин.
Кинины.
Серотонин.
Простагландины.
Экссудация с нарушением микроциркуляции и эмиграцией
Сосудистые реакции:
Ишемия - ангиоспастическая кратковременная (из-за разрушения адреналина и норадреналина под влиянием МАО и КОМТ), возможна обтурационная вследствие тромбоза.
Артериальная гиперемия обуславливает развитие одного из классических признаков воспаления (rubor) - красноты. Возможны все механизмы артериальной гипеперемии.
Смешанная гиперемия.
Венозная гиперемия – повышение проницаемости капилляров скопление жидкости в интерстиции сдавление венул и лимфатических капилляров. Классический признак - отек (tumor). Последствия:
Сгущение крови и повышение вязкости.
Активация гемостаза, адгезия и агрегация форменных элементов (монетные столбики, сладж), образование микротромбов, изменение физико-химических свойств крови, появление в крови некоторых белков (глобулины острой фазы), понижение альбумин-глобулинового коэффициента.
Маргинация (краевое стояние) лейкоцитов.
Набухание эндотелиальных клеток.
Стаз способствует гиперкоагуляции и тромбозу; возможны все виды стаза.
Собственно экссудация
Экстравазация жидкости из-за увеличения проницаемости сосудистой стенки. Другими словами происходит:
Разрушение стенки сосудов при альтерации.
Округление эндотелиальных клеток и появление межклеточных щелей (гистамин, брадикинин).
Микровизикуляция эндотелия - эндоцитоз и трансцитоз компонентов плазмы.
Раздвигание эндотелиальных клеток лейкоцитами по типу “расстёгивание молнии”.
Увеличение фильтрационного давления и площади фильтрации.
Различают раннюю экссудацию, 5-30 мин. (действие биогенных аминов и ацетилхолина на посткапиллярные венулы) и позднюю экссудацию, от 1 часа до 7 суток (действие полипептидных и липидных медиаторов на венулы и капилляры). Также происходит выход форменных элементов. Отличием экссудата от транссудата является наличие более 2-3% белка.
Эмиграция
Экстравазация форменных элементов - маргинация лейкоцитов, которая объясняется
изменением заряда поврежденных клеток,
фиксацией в межэндотелиальных щелях - “ловушках”,
движением с током жидкости,
образованием мостиков,
влиянием медиаторов (фибронектин, компоненты системы комплемента, XII фактор, каллекреин, брадикинин).
Движение лейкоцитов через сосудистую стенку. Лимфоциты и моноциты проникают через эндотелиальные клетки, не повреждая их. Полиморфноядерные лейкоциты - через эндотелиальные щели.
Движение клеток из сосуда в очаг воспаления по градиенту хемотаксинов называется хемотаксисом, в случае если это движение без градиента и беспорядочно - хемокинез.
Способность привлекать в очаг воспаления лейкоциты называется хемоаттракцией, ею обладают хемоаттрактанты:
Различные цитокины.
Микроорганизмы и их продукты.
Система комплемента и др. компоненты контактной системы.
Некротаксины - деграданты коллагена, фибронектин (гной лейкоцитов).
Иммунные комплексы, некоторые медиаторы (гистамин для эозинофилов).
Хемоаттрактанты воспринимаются рецепторами лейкоцитов.
Движение лейкоцитов обеспечивается структурами цитоскелета: микрофиламетами и микротрубочками. При участии Са++ и Са-связывающего белка гельзолина, актина цитоскелета. Желатинизация актина сопряжена с сокращением элементов цитоскелета.
Виды экссудатов:
Серозный (на слизистой - катаральный).
Фибринозный (крупозный и дифтерический на слизистой ротовой полости).
Гнойный.
Гнилостный.
Геморрагический (из-за анаэробов).
Хилёзный (за счёт жира из лимфатической системы брюшной полости).
28. – Фагоцитоз. Стадии фагоцитоза. Опсонизация – сущность, механизмы и значение.
Фагоцитоз (иммунные комплексы, липопротеиды, продукты распада коллагена, фибрина, бактерии).
Нейтрофилы (гранулоциты, микрофаги). Содержат гранулы, состоящие из лизосом и нелизосомальных ферментов (лизоцим, щелочная фосфагаза).
Мононуклеарные фагоциты (подвижные - моноциты, макрофаги и седлые (фиксированные)).
Стадии фагоцитоза
Приближение (случайное и хемотаксис).
Основныее медиаторы хемотаксиса
Интерлейкин 8.
С5а.
Лейкотриен В4.
Иммунные комплексы.
Фактор адгезии тромбоцитов.
Некротаксин.
Продукты микроорганизмов.
Контакт, распознавание и прилипание.
В процессе распознавания большую роль играет опсонизация - это покрытие объекта фагоцитоза сыворочными факторами - опсонинами (антителами IgG, М и Е, они «метят» объекты, подлежащие элиминации).
Прилипание осуществляется посредством связи опсонинов с рецепторами фагоцитов. Завершенный фагоцитоз идет только с участием опсонинов.
Поглощение (механизм: псевдоподии фагосомы фаголизосомы).
При поглощениее живых микроорганизмов,последние сначала должны быть убиты. В лейкоцитах существует 2 бактерицидных механизма:
зависящий от кислорода;
независящий от кислорода.
Зависящий от кислорода бактерицидный фактор связан с образованием активных метаболитов кислорода. Продукция этих веществ начинается после контакта фагоцитов с опсонизированными бактериями. Именно в это время фагоциты, которые в обычных условиях используют энергию анаэробного гликолиза, начинают усиленно поглощать кислород, что обозначают термином респираторный взрыв.
Возникновение его обусловлено активацией цитопламатической НАДФН-оксидазы, которая катализирует одноэлектронное восстановление молекулы кислорода до супероксидного радикального аниона, «отбирая» электрон от восстановленного пиридинового нуклеотида НАДФН:
оксидаза
2О2 + НАДФН 2О2- + НАДФ+ + Н+.
Расходуемые во время «респираторного взрыва» запасы НАДФН начинают немедленно восполняться усиленным окислением глюкозы через гексозомонофосфатный шунт.
Большая часть образующихся при восстановлении О2 супероксидных анионов О2- подвергается дисмутации до Н2О2:
2О2- + 2Н+ О2 + Н2О2.
Некоторая часть молекул Н2О2 взаимодействует в присутствии железа или меди с супероксидным анионом с образованием чрезвычайно активного гидроксильного радикала ОН:
О2- + Н2О2 ОН + ОН- + О2.
Цитоплазматическая НАДФН-оксидаза активируется в месте контакта фагоцита с микробом, а образование супероксидных анионов происходит на внешней стороне мембраны лейкоцитов, вне внутренней среды клетки. Процесс продолжается и после завершения образования фагосомы, вследствие чего внутри нее создается высокая концентрация бактерицидных радикалов. Проникающие внутрь цитоплазмы фагоцита радикалы нейтрализуются ферментами супероксиддисмутазой и каталазой.
Система образования бактерицидных метаболитов кислорода действует во всех профессиональных фагоцитах. В нейтрофилах совместно с ней действует еще одна мощная бактерицидная система – система миелопероксидазы (сходная с ней пероксидазная система имеется также у эозинофилов, но ее нет у моноцитов и макрофагов).
Миелопероксидаза - фермент, содержащийся в азурофильных гранулах нейтрофилов, катализирует реакцию между ионом галогена (обычно хлора) и перекисью водорода, что приводит к образованию хлорноватистой кислоты (гипохлоритного аниона ОС1-):
миелопероксидаза
CI- + Н2О2 OCI- + Н2О.
Гипохлорит оказывает выраженное бактерицидное действие сам по себе. Кроме того, он может реагировать с аммонием или аминами, образуя бактерицидные хлорамины.
Независящий от кислорода бактерицидный механизм связан с дегрануляцией - поступлением внутрь фагосомы бактерицидных веществ, которые содержатся во внутриклеточных гранулах фагоцитов.
Когда образование фагосомы завершается, к ней вплотную приближаются гранулы цитоплазмы фагоцитов. Мембрана гранул сливается с мембраной фагосомы, и содержимое гранул вливается внутрь фагосомы. Полагают, что стимулом к дегрануляции является увеличение цитозольного Са2+, концентрация которого возрастает особенно сильно вблизи фагосомы, где располагаются органеллы, накапливающие кальций.
Цитоплазматические гранулы всех облигатных фагоцитов содержат большое количество биологически активных веществ, способных убивать и переваривать микроорганизмы и другие поглощенные фагоцитами объекты. В нейтрофилах, например, имеется 3 типа гранул:
секреторные пузырьки;
первичные (азурофильные);
вторичные (специфические) гранулы.
Наиболее легко мобилизуемые секреторные пузырьки облегчают выход нейтрофилов из сосудов, их миграцию в тканях. Уничтожают и разрушают поглощенные частицы вещества азурофильных и специфических гранул. В азурофильных гранулах, помимо уже упомянутой миелопероксидазы, содержатся действующие независимо от кислорода низкомолекулярные бактерицидные пептиды дефенсины, слабое бактерицидное вещество лизоцим и множество разрушающих ферментов; в специфических гранулах лизоцим и белки, останавливающие размножение микроорганизмов, в частности, лактоферрин, связывающий необходимое для жизнедеятельности микроорганизмов железо.
На внутренней мембране специфических и азурофильных гранул находится протонный насос, который переносит водородные ионы из цитоплазмы фагоцита внутрь фагосомы. В результате рН среды в фагосоме понижается до 4-5, что вызывает гибель многих находящихся внутри фагосомы микроорганизмов. После того как микроорганизмы погибают, они разрушаются внутри фагосомы с помощью кислых гидролаз азурофильных гранул.
К числу важных бактерицидных факторов, действующих в активированных макрофагах, следует отнести и продукцию оксида азота (NO), которая осуществляется с помощью индуцибильной NO-синтазы. Фермент этот активируется -интерфероном, фактором некроза опухолей, ИЛ-1 и другими воспалительными цитокинами. NO действует цитостатически на опухолевые клетки, бактерии, паразиты, вирусы, ингибируя активность многих ферментов, участвующих в синтезе белков и нуклеиновых кислот. Оксид азота может соединяться с О2-, образуя пероксинитрит, который распадается на цитотоксические свободные радикалы ОН и NO-.
Переваривание за счёт сильных эндогенных окислителей и ферментов, таких как гидролазы, комплемент, лизоцим, аргиназа.
29. – Пролиферация. Роль трефонов. Особенности хронического воспаления. Понятия гранулемы.
Пролиферация - размножение клеток. Репаративная стадия воспаления. Нейтрофилы погибают, макрофаги расчищают поле для регенерации.
Фибробласты - главные эффекторы репарации. Механизм - стимуляция пролиферации через синтез ДНК и митотическую активность.
Медиаторы (трефоны)
Полиамины (иутресцин, спермидин, спермин) митогенный эффект.
Факторы роста фибробластов (тромбоцитарный и гипофизарный) ДНК, митоз.
Эндотелиальный хемотаксический фактор (из макрофагов) - индуцирует направленный рост сосудов в грануляционную ткань.
Кейлоны (ингибиторы пролиферации) уменьшение продукции - усиление пролиферации
Тканеспецифические стимуляторы пролиферации - иммуноглобулины G, М, антикейлон, -фетопротеин
Лимфокины (из Т - лимфоцитов). Пролиферация лимфоцитов и макрофагов.
Монокины (из макрофагов).
Ингибаторы пролиферации (тимидин, ПГ)
Нейротрофогены. Денервация.
СТГ, соматомедин, инсулин
Ослабляет: адреналин, глюкокортикоиды.
Усиливает: адреналин, альдостерон.
Хроническое воспаление – гранулёма.
Причина- слабая реактивность, чаще у детей и стариков, слабые сосудистые реакции. Незавершённый фагоцитоз. Помимо вышеназванных факторов – устойчивые микроорганизмы (при туберкулёзе, проказе, листериозе, токсикоплазмозе). Не перевариваемые объекты- инородные тела (металл, древесина). В эксперименте: декстран, коррагенан, зимозан, не перевариваемые полисахариды.
Суть - длительно раздражённые макрофаги выделяют монокины, которые и формируют гранулёму. Скопление мононуклеаров, гигантских клеток и соединительно тканных элементов формируют грануляционный вал.
Активация лимфоцитов как хронический аутоиммунный процесс бывает при ревматойдном артрите, системной красной волчанке. Этому способствует изменение антигенной структуры клеток в очаге воспаления.
30. – Лихорадка. Этиология. Пирогенны и их виды. Роль гипоталамуса в механизме развития лихорадки. Стадии и типы лихорадки.
Лихорадка – типовой патологический процесс, характеризующийся изменением терморегуляции и повышением температуры тела в ответ на действие пирогенных веществ.
Лихорадка - эволюционно выработанная приспособительная по своей основе реакция аппарата терморегуляции высших гомойтермных животных и человека на высокомолекулярные раздражители (пирогены) инфекционной природы и связанная с повреждением ткани, характеризующаяся временной перестройкой регуляции теплообмена на поддержание более высокого уровня температуры внутренней среды организма (П.Н. Веселкин).
В основе лихорадки лежит процесс перестройки терморегуляции, направленный на поддержание более высокой температуры тела. Поэтому к лихорадке нельзя относить иные случаи повышения температуры (все прочие виды гиепертермий), развившиеся вне связи с перестройкой центра терморегуляции на поддержание повышенной температуры.
Лихорадка относится к гипертермиям и имеет особое значение в патологии, т. к. сопровождает и участвует в сано- и патогенезе многих патологических процессов (воспаление, некоторые виды аллергии, гемолиз, введение сывороток и др.). Возникновение всех остальных гипертермий, помимо лихорадки, не связаны с действием пирогенов.
Классификация гипертермий
Пирогенно обусловленная (лихорадка).
Непирогенно обусловленные:
эндогенные гипертермии (психогенные, центрогенные, рефлексогенные, эндокринные, «злокачественная гипертермия»);
экзогенные гипертермии (перегревание, лекарственная);
поведенческая (при интенсивной физической нагрузке).
Этиология лихорадки
Экзогенные (инфекционные и неинфекционные) агенты.
Эндогенные (обломки клеток организма при травме, повреждении).
Непосредственная причина – попадание в организм пирогена.
Пирогенами (жаронесущими) называют такие вещества, которые, попадая в организм из вне или образуясь внутри него, вызывают лихорадку.
Первичные пирогены – чаще всего липополисахариды и липоид А клеточных мембран грамм-отрицательных микроорганизмов, могут быть так же белки и нуклеиновые кислоты.
Учитывая положительное значение повышенной температуры для организма, существуют лекарственные препараты не для снижения, а для повышения температуры организма (пирогенал). Пиротерапия применяется для ускорения скорости всех химических реакций и активации вследствие этого большинства органов и систем с целью активации иммунитета (синтез интерферона, лизоцима, интерлейкинов, фактора некроза опухолей, антител, усиление фагоцитоза) при хронических инфекционных и опухолевых заболеваниях, повышения детоксикационных функций организма, искусственного обострения вялотекущих, хронических воспалений.
Вторичные пирогены - образуются (синтезируются) в фагоцитах (нейтрофилах, моноцитах, тканевых макрофагах) при фагоцитозе токсинов, имеют пептидную природу. К ним относятся интерлейкины 1 и 6 (ИЛ1, ИЛ6), фактор некроза опухолей (ФНО), интерферон.
Стадии лихорадочного процесса:
Подъем температуры.
В первую очередь резко уменьшается теплоотдача. Активация симпатоадреналовой системы приводит к снижению кровообращения периферических отделов организма - кожные покровы бледные (возбуждение -адренорецепторов). Возбуждение -адренорецепторов сопровождается учащением дыхания и сердцебиений, увеличением скорости кровотока (из-за усиления работы сердца). Увеличение частоты сердечных сокращений обусловлено также повышением возбудимости синусового узла при повышении температуры «ядра».
Симпатоадреналовая реакция сопровождается увеличением АД, а в следствие этого растет фильтрационное давление в почках и увеличивается диурез.
Возбуждение -адренорецепторов бурой жировой ткани также сопровождается увеличением теплопродукции.
Активация холодовых рецепторов вызывает возбуждение центра теплопродукции начинается процесс мышечного сокращения - озноб; мышечная дрожь.
Выработка тиролиберина в гипоталамусе сопровождается увеличением продукции ТТГ в щитовидной железе (разобщение окисления и фосфорилирования).
Организм начинает внутренне согреваться и не отдает тепло. Теплопродукция превышает теплоотдачу. Причем в начале это происходит за счет уменьшения теплоотдачи. Температура тела начинает увеличиваться.
Стояние температуры.
Температура стабилизируется на постоянном высоком уровне. Центр терморегуляции, перейдя на другой уровень, подключает терморегуляцию за счет увеличения теплоотдачи, причем теплоотдача увеличивается ровно на столько, чтобы уравновесить теплопродукцию при повышенной температуре. На этой стадии наблюдаются сосудистые реакции: расширение периферических сосудов, в результате чего бледность сменяется гиперемией. Больные реагируют в этой стадии на тепло и холод соответствующими реакциями, как и здоровые.
АД постоянное, а усиление секреции альдостерона вызывает снижение диуреза (смысл – разбавление возбудителя).
Снижение температуры (разрешение).
Центр терморегуляции возвращается к своему прежнему уровню активности. Нормализуется функция симпатической НС. Теплоотдача преобладает над теплопродукцией за счет потоотделения (активация парасимпатической НС). Наблюдается снижение температуры. АД снижается, а диурез возрастает.
Если температура падает слишком быстро (критически), развивается сосудистая недостаточность - коллапс. Наблюдается дефицит объема циркулирующей крови по отношению к объему резко расширенного сосудистого русла. Поэтому легче переносится больным постепенное снижение температуры (литическое).
Информация о состоянии терморегуляции анализируется в гипоталамусе, где суммируются все термические сигналы. Передняя часть центра терморегуляции отвечает за теплоотдачу, задняя - за теплопродукцию, т.е. химическую терморегуляцию.
излучение
(радиация) конвекция испарение кондукция несократительный
термогенез сократительный
термогенез
Гипоталамус
Теплоотдача
Теплопродукция
Разобщение окислительного фосфорилирования
31. – Ответ острой фазы. Влияние лихорадки на организм. Положительное и отрицательное значение лихорадки. Сходство и отличия лихорадки от перегревания.
Обмен веществ при лихорадке
Белковый обмен
Введение пирогенных веществ не вызывает нарушений белкового обмена. В то же время при заболеваниях, сопровождающихся лихорадкой, часто обнаруживается отрицательный азотистый балланс - увеличивается выделение с мочой азотистых продуктов обмена, в частности мочевины и мочевой кислоты. Однако параллелизма между усилением распада белка и высотой температуры часто не наблюдается, и значительное усиление катаболизма обычно связано с инфекционной интоксикацией, в том числе с ее отрицательным влиянием на пищеварительную систему.
Причиной распада белка при ряде инфекционных заболеваний является интоксикация, дегенеративные и воспалительные изменения в тканях, а также сопутствующее голодание из-за пониженного аппетита и ухудшения усвоения пищи. Потеря аппетита связана с высоким уровнем ИЛ1; снижение массы тела - как с анорексией, так и с преобладанием катаболических процессов над анаболическими под действием ИЛ1 и фактора некроза опухоли. Они также снижают активность липопротеинкиназы, блокируя тем самым неолипогенез в жировой клетчатке.
Углеводный обмен
Изменения в углеводном обмене происходят за счет активации гипоталамо-гипофизарно-надпочечниковой системы. В печени активируется гликогенолиз, снижаются запасы гликогена, в крови отмечается гипергликемия. Содержание гликогена в сердце не изменяется.
Жировой обмен
Изменения в жировом обмене происходят за счет активации гипоталамо-гипофизарно-надпочечниковой системы. При уменьшении углеводных резервов отмечается усиленное использование жиров для теплопродукции. При истощении в печени запасов гликогена окисление жира идет не до конечных продуктов, накапливаются кетоновые тела, с мочой выделяется ацетон (ацетонурия). Ацетонурия наблюдается лишь в случаях нарушенного питания больного, когда голодание способствует мобилизации жира из жировых депо.
ИЛ1 и фактора некроза опухоли снижают активность липопротеинкиназы, блокируя тем самым неолипогенез в жировой клетчатке.
Водно-электролитный обмен
В первой стадии усиление почечного кровотока сопровождается повышением диуреза. Во второй стадии диурез снижается, происходит задержка воды в ряде органов, мышцах и воспалительных очагах (в виде экссудата). В связи с повышенной секрецией альдостерона ограничивается выведение из организма ионов натрия. Вместе с натрием задерживаются ионы хлора. В третьей стадии лихорадки диурез повышается, увеличивается секреция воды потовыми железами, с мочой и потом выделяется значительное количество хлорида натрия.
Изменение функции органов при лихорадке
Центральная нервная система
Наиболее частыми жалобами у лихорадящих больных являются головная боль, сонливость, разбитость, апатия, гиперестезия. Механизм этих проявлений связывают с эффектами ИЛ1. Могут быть бред, галлюцинации. Заболевания, сопровождающиеся лихорадкой, могут протекать как с явлениями угнетения высшей нервной деятельности, так и ее активации. Очищенные бактериальные пирогены обычно не оказывают отрицательного влияния на ЦНС.
Сердечно-сосудистая система
При лихорадке, как правило, увеличиваются частота и сила сердечных сокращений, что приводит к увеличению ударного и минутного объема. Повышение температуры на 1°С сопровождается обычно учащением сердцебиения на 8-10 ударов в 1 мин, которое обусловлено повышением тонуса симпатического отдела вегетативной нервной системы и прямым действием нагретой крови на синусовый узел сердца.
Длительная непрерывная гипердинамия сердца, особенно при высокой лихорадке и ранее пораженном каким-либо патологическим процессом сердце (например, в результате инфаркта миокарда), может привести к перегрузочной форме его недостаточности.
Артериальное давление в начале лихорадки несколько повышено за счет спазма периферических сосудов. Приток крови к внутренним органам увеличивается за счет ограничения периферического кровообращения. На этом, в частности, основана пиротерапия почечной гипертонии. В период критического падения температуры давление может резко снизиться из-за ослабления тонуса сосудов.
Система дыхания
В первой стадии лихорадки частота дыхания незначительно снижается. Во второй стадии частота дыхательных движений увеличивается, иногда в два-три раза, но легочная вентиляция при этом практически не изменяется, так как глубина дыхания уменьшается. Отмечено, что повышение температуры головного мозга вызывает учащенное дыхание (тахипноэ). Вместе с тем, потребление кислорода тканями мозга в диапазоне изменений температуры 38-42°С нарастает незначительно.
Система пищеварения
Одной из постоянных жалоб больных с лихорадочными заболеваниями является потеря аппетита. У них происходит снижение секреции слюны, язык становится сухим, обложенным. Снижается секреторная деятельность всех пищеварительных желез. Отмечаются также двигательные расстройства желудка и кишечника. Преобладание возбуждения симпатического или парасимпатического отдела нервной системы в разные стадии лихорадки приводит к изменению тонуса кишечника, возникают спастические или атонические запоры.
Защитные реакции при лихорадке
При лихорадке активно увеличиваются процессы антителообразования. Стимулируется иммунитет.
Это особенно важно, потому что, например, в 60-е годы, когда начали интенсивно применять жаропонижающие средства появились случаи повторного заболевания инфекциями, к которым при обычно лихорадке образовывался стойкий иммунитет (корь, например).
Активизация фагоцитоза.
Увеличивается ферментативная активность, обеспечивается энергетическая эффективность различных процессов при минимальных расходах энергии.
Повышается дезинтоксикационная функция органов (в том числе печени).
Подавляется активность возбудителей инфекции.
Активируется стрессовая реакция - развивается общий адаптационный синдром.
Отрицательное значение лихорадки
Истощение энергетических ресурсов.
Гипоксия и ацидоз.
Дисфункция сердечно-сосудистой системы.
Угнетение центральной нервной системы.
Лихорадка и перегревание: сходства и различия.
Перегревание - реакция аппарата терморегуляции, не имеет системного характера, и повышение температуры обычно связано с влиянием на отдельные звенья терморегуляции или непосредственно на обмен веществ в тканях.
Отличительные признаки перегревания организма и лихорадки
Признак |
Перегревание организма |
Лихорадка |
Этиология |
Уменьшение теплоотдачи. Травмы, кровоизлияния, опухоли, инфекции головного мозга. Химические соединения, нарушающие сопряженность между свободным дыханием и фосфорилированием. |
Воздействие пирогенных веществ на центр терморегуляции. |
Патогенез |
Формирование функциональной системы, поддерживающей гипертермию при действии высокой температуры окружающей среды или факторов, затрудняющих термолиз. |
Функциональная перестройка ценра терморегуляции, наблюдающаяся в результате действия пирогенных веществ. |
Состояние терморегуляции |
Температура тела начинает повышаться после того, как теплоотдача оказывается меньше теплопродукции. |
Температура тела активно регулируется на новом уровне. |
Направленность изменений терморегуляции |
На термолиз. |
На термогенез. |
Чем определяется степень повышения температуры |
Физическими условиями теплообмена организма с окружающей средой. |
Уровнем смещения «установочной точки» в центре терморегуляции. |
Озноб |
Отсутствует. |
Отмечается в I стадии. |
Усиленное потоотделение |
Как правило, есть. |
Отмечается в III стадии. |
Эффективность жаропонижающей терапии |
Не эффективна. |
Эффективна. |
Другие виды гипертермий, помимо лихорадки, называют перегреванием. Это ряд эндогенных гипертермии, имеющих черты сходства с лихорадкой, однако не связанных с действием пирогенов. Существуют определенные разногласия по вопросу о правомерности отнесения таких состояний к лихорадкам. Поэтому для их обозначения будет использован термин «лихорадоподобные состояния» (ЛПС).
Неврогенные ЛПС подразделяют на центрогенные, психогенные и рефлексогенные.
Центрогенные ЛПС могут возникать при повреждении различных отделов головного мозга (кровоизлияниях, опухолях, травме, отеке мозга и т.п.). Причиной развития психогенных ЛПС могут быть функциональные нарушения высшей нервной деятельности (невроз, психические расстройства), значительное эмоционально-умственное напряжение; описаны случаи возникновения гипертермии под влиянием гипнотического внушения.
Рефлексогенные ЛПС могут наблюдаться при почечнокаменной болезни, желчнокаменной болезни, раздражении брюшины, катетеризации уретры и т.д. При этом, как правило, возникает болевой синдром; нельзя исключить в некоторых случаях и микроповреждения тканей, приводящего к образованию первичных неинфекционных пирогенов. В таких случаях, очевидно, наряду с рефлекторным механизмом будет участвовать и обычный механизм действия пирогенных веществ.
Эндокринные ЛПС наблюдаются при некоторых эндокринопатиях, с особенным постоянством при гипертиреозах.
Лекарственные ЛПС возникают при энтеральном или парентеральном введении некоторых фармакологических препаратов: кофеина, эфедрина, метиленовой сини, гиперосмолярных растворов и др.
32. – Иммунодефицитные состояния. Первичные и вторичные иммунодефициты. СПИД- этиология и патогенез.
Иммунодефицитные состояния: первичные и вторичные. Патогенез СПИДа.
Иммунодефициты - нарушения нормального иммунологического статуса, которые обусловлены дефектом одного или нескольких механизмов иммунного ответа. Они подразделяются на первичные (врождённые и генетически обусловленные) и вторичные.
Вторичные иммунодефициты – это недостаточность иммунной системы, возникшая как следствие какого–то заболевания у ранее здорового человека, эта недостаточность может быть обратимой при устранении вызвавшей её причины.
Вторичный иммунодефицит встречается значительно чаще, чем первичный, почти каждое серьёзное заболевание с длительным течением в какой–то степени ослабляет иммунную систему, а также различные виды лечения химио- и лучевая терапия; все без исключения препараты, которые используют для лечения сердечно-сосудистой системы, ЖКТ, нервной системы оказывают седативное воздействие и на иммунные клетки.
Одним из наиболее клинически значимых вторичных иммунодефицитов является синдром приобретенного иммунодефицита (СПИД). Следует отметить, что термином СПИД обозначается лишь последняя стадия (по клиническому течению) ВИЧ-инфекции, поскольку иммунодефицит формируется только на заключительном этапе болезни. Т.е. правильное название заболевания именно ВИЧ-инфекция (инфекция вирусом иммунодефицита человека).
Впервые синдром описан в научной литературе в 1981 г. американскими исследователями. Однако ретроспективный анализ позволяет утверждать, что СПИД поражал людей и ранее. Первые случаи синдрома официально были зарегистрированы в США, Африке, на Гаити. В последние годы, когда были налажены методы диагностики СПИДа, выяснилось, что каждые 12-14 месяцев число зарегистрированных случаев синдрома удваивается. Соотношение инфицированных лиц к заболевшим колеблется от 50:1 до 100:1.
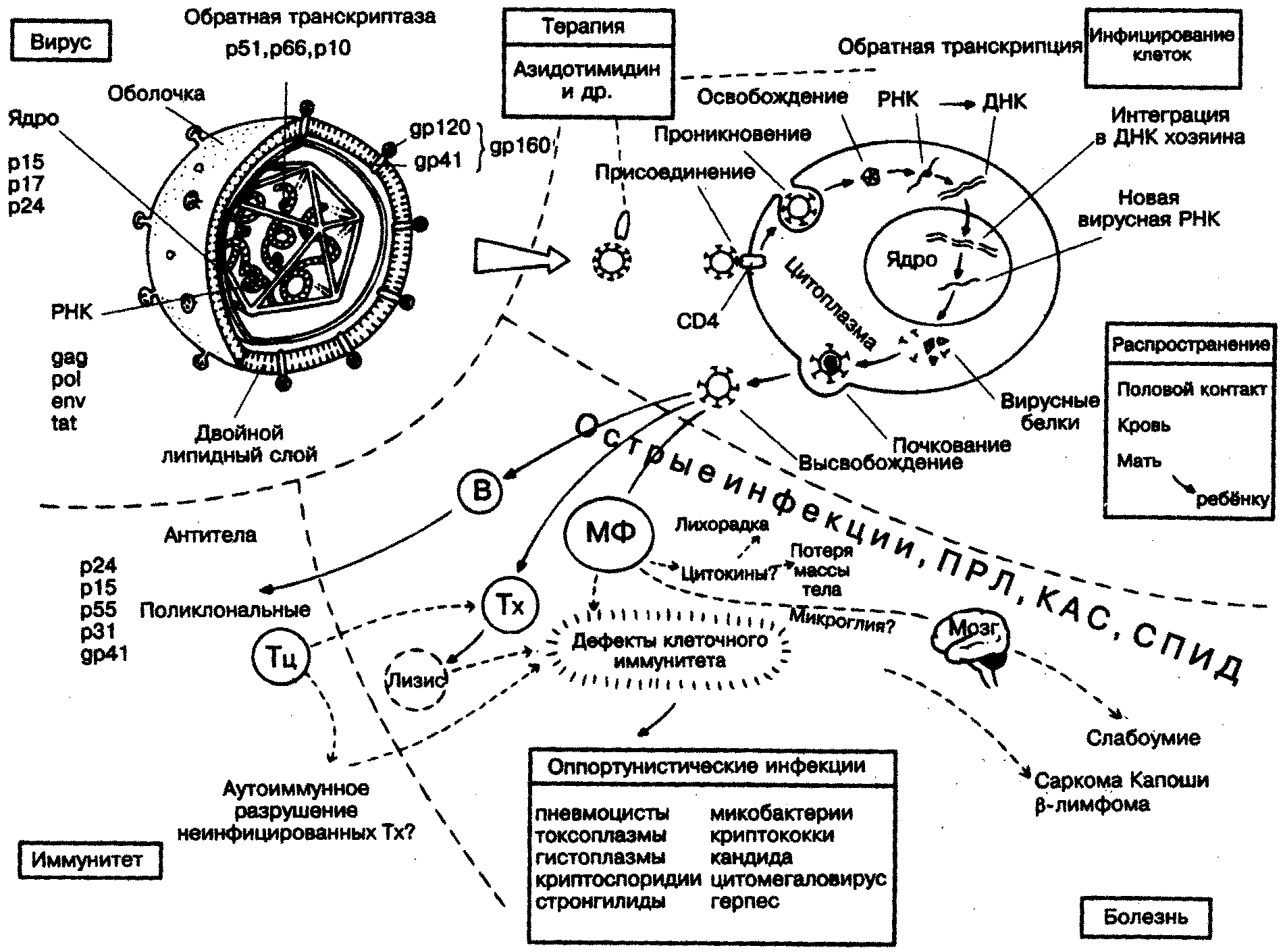
Рис. 12.5. Схема развития и проявлений ВИЧ-инфекции и СПИДа.
Наибольшее распространение ВИЧ-инфекция имеет среди гомо- и бисексуальных мужчин, наркоманов, вводящих наркотики внутривенно и пользующихся "коллективными" шприцами; реципиентов гемотрансфузий (больные анемиями); детей родителей, больных ВИЧ-инфекцией.
Возбудитель ВИЧ-инфекции относится к группе ретровирусов подсемейства лентивирусов. Они содержат однонитчатую линейную РНК и фермент ревертазу (РНК-зависимая ДНК полимеразу). Репликация вирусной нуклеиновой кислоты идет через стадию синтеза двунитчатой ДНК на матрице РНК, т.е. как бы обратным путем. В клетку-мишень проникает ДНК-копия с РНК вируса, котрая интегрируется с клеточным геномом. Транскрипция информации вирусной ДНК осуществляется при участии клеточной РНК-полимеразы. Созревание вириона путем почкования идет на клеточных мембранах. В организм вирус проникает с кровью и ее дериватами, клетками при пересадке тканей и органов, переливании крови, со спермой и слюной через поврежденную слизистую или кожу. Проникнув в организм, возбудитель ВИЧ-инфекции внедряется в клетки, имеющие рецепторы CD4, к которым гликопротеиды вирусной оболочки имеют высокий аффинитет. Наиболее богаты рецепторами CD4 Т-лимфоциты-хелперы, в которые в основном и проникают вирусы. Однако помимо этого вирус способен внедряться и в моноциты, фагоцитирующие клетки, нейроны, клетки нейроглии, слизистой прямой кишки.
Вирус обнаруживается в крови, в ткани слюнных желез, простаты, яичек. Через 6-8 недель (реже - через 8-9 месяцев) после инфицирования появляются антитела к ВИЧ.
Патогенез вич-инфекции
ВИЧ, инкорпорированный в геноме клеток организма в форме провируса, стимулирует транскрипцию РНК-вируса. На основе этой РНК синтезируются белковые компоненты вируса, которые затем интегрируют с его нуклеиновой кислотой. По завершении процесса "сборки" вирусные частицы отторгаются от клетки, попадают в межклеточную жидкость, лимфу, кровь и атакуют новые клетки, имеющие рецепторы CD4, приводя их к гибели.
Существует несколько версий о механизме лизиса клеток, пораженных ВИЧ.
Одно из допущений (Р.Галло,1983) заключается в разрушении мембран лимфоцитов, моноцитов, нейронов при "отпочковывании" вируса от клетки с последующим их лизисом. Вероятность гибели клеток пропорциональна количеству рецепторов CD4 на их поверхности. Наибольшее их число имеют Т-хелперы, в связи, с чем их количество значительно уменьшается.
В качестве другого механизма лизиса инфицированных ВИЧ клеток рассматривается возможность встраивания белков вирусной оболочки в клеточные мембраны. В связи с этим клетки распознаются ИКС как чужеродные и уничтожаются (Р.Курт, Х.Бреде,1984).
Гибель клеток связана не только с непосредственным цитопатогенным действием вируса, но и с индукцией апоптоза клеток вследствие перекрестного связывания CD4 белком gp 120.
Полагают также, что инкорпорация ДНК вируса (провируса) в геном Т-хелпера лишает их способности к трансформации и реагированию на регуляторные стимулы, в частности - на ИЛ2.
Допускается также регуляторное подавление Т-хелперов растворимыми факторами супрессии, которые выделяют мононуклеары крови больных СПИДом (Дж. Лоуренс,1983).
Эти и другие механизмы действия ВИЧ на клетки обуславливают их лизис, вызывая уменьшение их числа. В наибольшей мере это относится к Т-хелперам. Именно по этому развивается лимфопения. Кроме того, подавляется способность Т-хелперов продуцировать ИЛ2.
Одновременно наблюдается снижение (примерно на 80-90%) количества и функциональной активности естественных киллеров (NK-клетки).
Число В-клеток, как правило, остается в пределах нормы, а функциональная активность их нередко снижается.
Количество макрофагов обычно не изменяется, однако выявляется нарушение хемотаксиса и внутриклеточного переваривания чужеродных агентов. Отмечается также расстройство механизма "презентации" макрофагом антигена Т- и В-лимфоцитам.
Указанные изменения создают предрасположенность больных СПИДом к инфекциям, лимфоретикулярным опухолям (например, к саркоме Капоши), а также - неспособности к развитию аллергических реакций замедленного типа.
Первичные иммунодефициты - такие состояния, при которых нарушение иммунных механизмов (продукции антител или Т-лимфоцитов) часто связано с генетическим блоком.
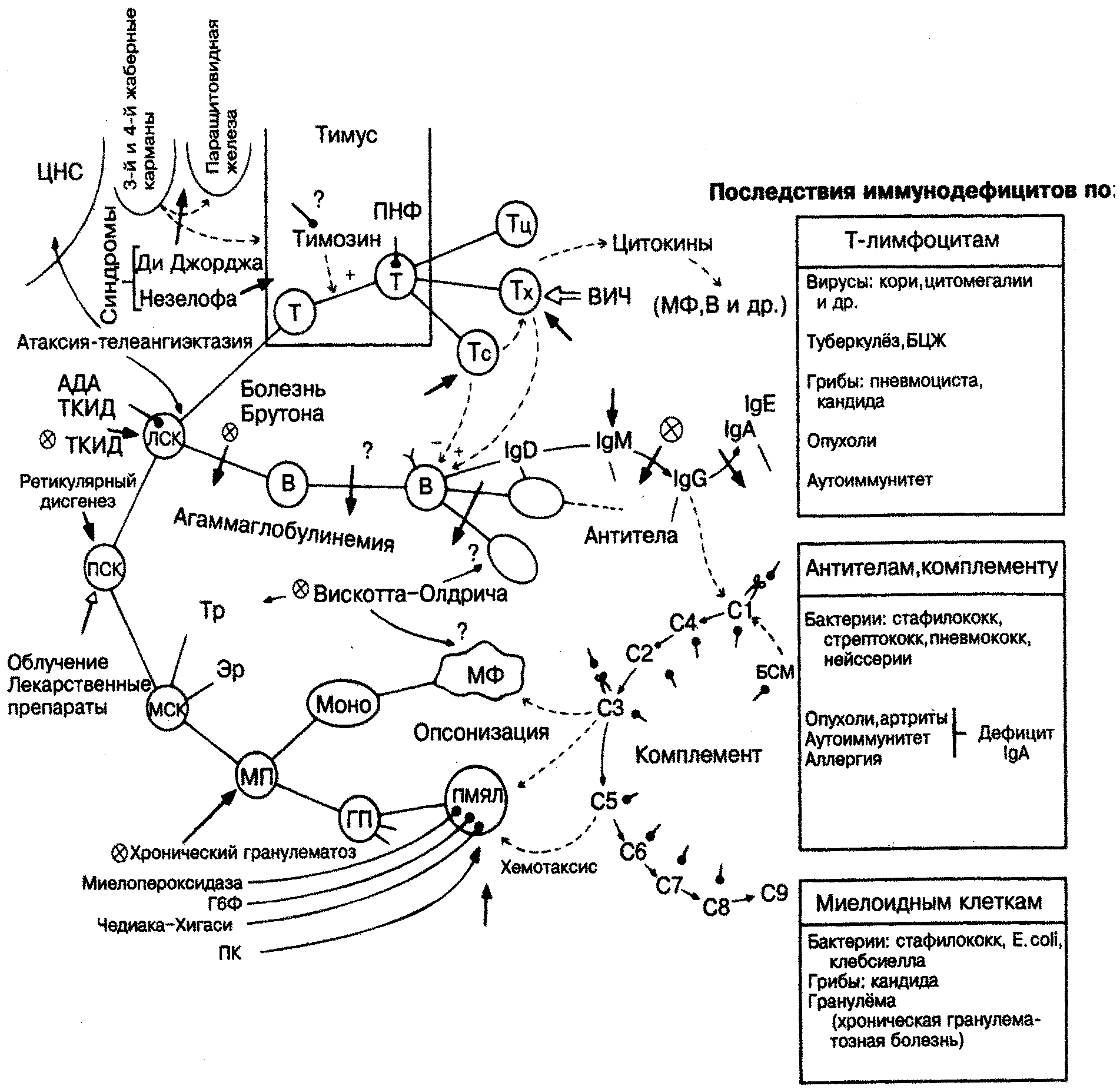
Рис. 12.6. Общая схема иммунодефицитов.
Их подразделяют на 4 главные группы, в зависимости от того, какой компонент иммунной системы неполноценен: дефекты В– или Т– лимфоцитов, фагоцитирующих клеток или комплемента. Лидирует патология В–клеток, т. е. дефекты выработки антител, она составляет более 50% случаев первичного иммунодефицита, патология Т – клеток – 30%, дефекты системы фагоцитов менее 18%, недостаточность комплемента – приблизительно 2%, т. е. наиболее уязвимы те отделы иммунитета, которые эволюционно сформировались позднее.
Патология в–клеток
Агаммаглобулинэмия характеризуется отсутствием В-клеток (болезнь Брутона), неспособностью В–лимфоцитов дифференцироваться в плазматические клетки.
Гипогаммаглобулинэмия или избирательная неспособность к продукции какого–либо одного класса иммуноглобулинов, чаще Ig А – селективный имунодефицит Ig А, встречается 1:400. У этих больных часто развиваются аллергия и полиартрит. Страдают рецидивирующими бактериальными инфекциями: стафилококк, стрептококк, пневмококк, нейсерии.
Дефекты с преимущественным поражением т–клеток
Синдром Ди–Джорджи – гипоплазия тимуса и паращитовидных желез вследствие недоразвития 3-4 жаберных карманов (характерны гипокальциемическая тетания, врождённая патология сердца), рецидивирующие инфекции появляются уже вскоре после рождения. Инфекции для Т–иммунодефицита другие: вирусные: корь, цитомегаловирусные, туберкулёз, БЦЖ, грибы, пневмоцисты, кандидоз.
Тяжёлый комбинированный иммунодефицит ТКИД (дефекты Т и В–лимфоцитов). Выраженное функциональное нарушение гуморального и клеточного иммунитета. Имеются многообразные дефекты в развитии иммунокомпетентных клеток. Характерны выраженная лимфопения и агаммаглобулинемия. Болеют в основном дети, которые погибают в раннем возрасте.
ИД с атаксией–телеангиэктазией, синдром Вискотта–Олдрича - иммунный дефект проявляется в нарушении клеточного иммунитета и снижении антителообразования, имеется триада клинических признаков - экзема, тромбоцитопения и инфекционные осложнения.
Дефекты миелоидных клеток
Хроническая гранулёматозная болезнь – дефект генерации активных форм кислорода.
Синдром Чедиака–Хигаси – не формируют фаголизосомы, в других случаях нарушается хемотаксис – «ленивые лейкоциты».
Дефекты системы комплемента (возможен генетически обусловленный дефицит любого компонента комплемента)
Дефект инактиватора – например ингибитора С1 (наследственный ангионевротический отёк).
Дефекты С1,С4,С2 обуславливают предрасположенность к болезням иммунных комплексов (СКВ), С5–С9 – предрасположенность к инфекции, вызванной нейсериями, С3 – самые тяжёлые к летальному исходу.
34.- Аллергия. Аллергены и их виды. Стадии аллергических реакций. Понятие аутоиммунных заболеваний.
Понятие "аллергия" было впервые употреблено в 1906 г австрийским педиатром Clemens
von Pirguet для определения реактивности.
Сегодня под аллергией понимается форма иммунного ответа организма на вещества антигенной или гаптеновой природы, сопровождающаяся повреждением структуры и функции собственных клеток, тканей и органов.
Этиология аллергических заболеваний
Причиной аллергических заболеваний является аллерген, условиями их возникновения – определенные особенности внешней среды и состояние реактивности организма.
Аллерген вещество, вызывающее развитие аллергической реакции. Если введение вещества приводит к развитию аллергической реакции, то его называют аллергеном, если к развитию иммунной антигеном.
Следовательно, аллергены обладают всеми свойствами антигенов (макромолекулярность, преимущественно белковая природа, чужеродность для данного организма и др.).
Многие молекулярные соединения, например, лекарственные препараты, простые химические вещества (бром, йод, хром, никель и др.), а также более сложные продукты небелковой природы (некоторые микробные продукты, полисахариды и др.), их называют гаптенами, при попадании в организм, не включает иммунных механизмов, но становятся антигенами после соединения с белками тканей организма.
Однако не каждое соединение химического вещества с белком приводит к образованию антигена. Многие лекарственные препараты, эндогенно образующиеся микромолекулярные соединения (стероидные гормоны, ионы меди, железа, продукты метаболизма) соединяют с сыворочными белками, которые выполняют транспортную роль.
Все аллергены принято делить на две группы: экзо- и эндоаллергены (или аутоаллергены) образующиеся в самом организме.
Существует классификация, в основе которой лежит способ попадания аллергена в организм:
воздушные, ингаляционные аллергена (бытовая и производственная пыль, пыльца растений, эпидермис и шерсть животных и др.)
пищевые аллергены
контактные аллергены, проникающие через кожу и слизистые оболочки (химические вещества, лекарства)
инъекционные (сыворотки, лекарства)
инфекционные (бактерии, вирусы)
лекарственные. В каждую группу этой классификации входят аллергены различного происхождения.
Классификация экзогенных аллергенов по происхождению
аллергены неинфекционного происхождения: бытовые, эпидермальные, пыльцевые, пищевые, промышленные.
инфекционного происхождения: бактериальные, грибковые вирусные.
Домашняя пыль сложный по своему составу аллерген, в который входят пылевые частицы одежды, постельного белья, мебели, грибы (в сырых помещениях), домашние насекомые, бактерии; основной компонент – клещи вида Dermatophagoideus, обитающие в постелях, подушках, питаясь чешуйками рогового слоя эпидермиса.
Эпидермальные: перхоть, шерсть животных, перья птиц, чешуя рыб.
Лекарственные – любой за исключением раствора глюкозы, хлорида натрия и т. п.
Пыльцевые – пыльца не всех видов растений, а только достаточно мелкая (диаметр не более 30 мкм) и обладающая хорошими летучими свойствами. В каждой климатогеографической зоне свои виды растений. У нас в городе весной – береза (тополь не аллерген), осенью – полынь, в деревнях тимофеевка и др. луговые травы. В США – амброзия.
Различные виды пыльцы могут иметь общие аллергены (пыльца злаковых) и поэтому могут давать перекрестные реакции. Лицам, страдающим поллинозами, не стоит увлекаться применением внутрь лечебных трав.
Стадии развития аллергии
Иммунологическая - охватывает все изменения в иммунной системе, возникающие с момента поступления аллергена в организм, образования антител или сенсибилизированных лимфоцитов и соединения их с повторно поступившим или персистирующим в организме аллергеном.
Сенсибилизация – иммунологическое опосредованное повышение чувствительности к антигену, т. е. образование антител или сенсибилизиронных лимфоцитов.
Патохимическая, или стадия образования медиаторов - образование биологически активных веществ; стимулом к их возникновению является соединение аллергена с антителами или сенсибилизированных лимфоцитов в конце иммунологической стадии.
Патофизиологическая или стадия клинических проявлений - характеризуется патогенным действием образовавшихся медиаторов на клетки, органы и ткани организма.
Аутоиммунные (аутоаллергические) болезни представляют собой группы заболеваний, основным механизмом развития которых являются реакции сенсибилизированных лимфоцитов и аутоантител с тканями организма.
В роли аутоантигенов могут выступать:
естественные, первичные антигены (неизменная ткань хрусталика глаза, щитовидной железы, яичка, нервной ткани);
приобретенные, вторичные (патологически измененные ткани) антигены как инфекционной, так и не инфекционной природы.
Неинфекционные аутоантигены по происхождению своему могут быть ожоговым, лучевым, холодовым и др., а инфекционные - комплексными и промежуточными.
Появление естественных аутоантигенов связывают с нарушением физиологической изоляции органов и тканей, по отношению к которым отсутствует иммунологическая толерантность. Известно, что в период созревания лимфоидной ткани возникает иммунологическая толерантность к антигенам всех органов и тканей, кроме тканей глаза, щитовидной железы, семенников, надпочечников, головного мозга, нервов. Считается, что антигены этих органов и тканей отграничены от лимфоидной ткани гистогематическим барьером.
Приобретенные неинфекционные аутоантигены могут появляются при воздействии на ткани физических и химических факторов, под влиянием лекарственных препаратов. В образовании аутоантигенов в этих условиях большое значение отводится гаптенному механизму.
Появление инфекционного комплексного аутоантигена (комплексы ткань-микроб, ткань-токсин) ведет к тому, что возникающие в этих условиях аутоантитела реагируют ни только с микробом, но и с тканью, что и определяет возможность развития аутоагрессивного процесса.
Близкая ситуация возникает в том случае, когда появляются перекрестно реагирующие антигены, т.е. антигены микробов, имеющие общие детерминанты с антигенами ткани. Доказана, например, антигенная общность 5-го серотипа -гемолитического стрептококка и ткани почечных клубочков, клебсиеллы и ткани легких, некоторых штампов кишечной полочки и ткани слизистой оболочки толстой кишки и др.
Инфекционные промежуточные антигены (вирусиндуцированные), которые отличаются по своим антигенным свойствам как от клетки, так и от вируса, способны индуцировать продукцию антител и тем самым вызывать аутоиммунное повреждение клеток и тканей.
Однако основной причиной аутоиммунизации считают нарушения в центральном органе иммуногенеза - тимусе, которые приводят к потере способности естественных иммунодепрессантов подавлять функцию Т-клеток.
Классификация аутоиммунных болезней
Органоспецифические аутоиммунные болезни (болезнь Хашимото, энцефаломиелит, полиневрит, рассеяный склероз, идиопатическая аддисонова болезнь, асперматогения, симпатическая офтальмия). Их возникновение провоцирует инфекция, особенно вирусная, хроническое воспаление и др. Аутоиммунизация развивается в связи с повреждением физиологических барьеров иммунологически обособленных органов, что позволяет иммунной системе реагировать на их антигены выработкой аутоантител и сенсибилизированных лимфоцитов. При этом в органах развиваются изменения, характерные преимущественно для реакции ГЗТ.
Органонеспецифические аутоиммунные болезни (системная красная волчанка, ревматоидный артрит, системная склеродермия, дерматомиозит, вторичная гемолитическая анемия и тромбоцитопения). В этих случаях нарушения контроля иммунологического гемостаза лимфоидной системы связаны с генетическими факторами,вирусной и бактериальной инфекцией, ионизирующим излучением. Аутоиммунизация развивается к антигенам многих органов и тканей, не обладающих органной специфичностью. В органах и тканях при этих заболеваниях наблюдаются изменения, характерные для реакций как ГЗТ, так и особенно ГНТ.
Это определенные формы гломерулонефрита, гепатита, хронического гастрита и энтерита, неспецифический язвенный колит, цирроз печени, ожоговая болезнь, аллергические анемии, агранулоцитоз, лекарственная болезнь. Изменения антигенных свойств тканей и органов, т.е. образование аутоантигенов при этих заболеваниях, связано, прежде всего, с денатурацией тканевых белков при ожоге, травме, хроническом воспалении, вирусной инфекции. Образование аутоантигена возможно при воздействии бактериального антигена, особенно перекрестно реагирующего. В этих случаях с аутоиммунизацией связано не возникновение заболеваний, а прогрессирование характерных для него органных изменений, которые отражают реакции ГЗТ и ГНТ.
Аллергические реакции немедленного типа. Патогенез анафилактического шока.
Так как иммунный ответ может быть гуморальным и клеточным, так и в основе аллергических реакций могут участвовать как антитела, так и сенсибилизиронные Т–лимфоциты. Наибольшее распространение получила классификация, в которой выделены аллергические реакции немедленного типа (син.: гиперчувствительность немедленного типа) и аллергические реакции замедленного типа. В основу классификации положено время появления реакции после контакта с аллергеном. Реакции немедленного типа развивались в течении 15-20 мин. (обусловлены антителами), замедленного типа – через 1-2 суток (обусловлены лимфоцитами).
Классификация, предложенная P. Yell, R. Cooombs (1968), основана на патогенетическом принципе. В соответствии с этой классификацией выделено 4 типа иммунных реакций.
Таблица 12.1.
Типы аллергических реакций.
Тип |
Наименование типа реакции |
Иммунный механизм реакции |
1. |
Анафилактический |
Ig E и реже Ig G4 антитела |
2. |
Цитоксический |
Ig G и Ig M антитела |
3. |
Иммунокомплексный |
Ig G и Ig M антитела |
4. |
Замедленная гиперчувствительность |
Сенсабилизированные лимфоциты |
Выделяют еще 5 тип – рецепторноопосредованный или антирецепторный (иммунный тип сахарного диабета, иммунные заболевания щитовидной железы, гипофиза).
I. Реагиновый тип повреждения тканей или анафилактический. Связан с образованием особого типа антител цитотропных или цитофильных Ig E или G4, имеющих высокое сродство к определенным клеткам (тучными, базофилами). Антиген, вступая во взаимодействие с фиксированным на клетках антителами, приводит к секреции предсуществующих (гистамин, серотонин, эозинофильные хемотаксичные факторы) и вновь образущихся (простагландины, лейкотриены, ТАФ) медиаторов, это классический путь. Они действуют на сосуды и клетки – мишени, опосредованно включая в развитие аллергической реакции эозинофилы, нейтрофилы, тромбоциты, которые в свою очередь также начинают выделять медиаторы, обозначаемые как вторичные – фосфолипаза Д, арилсульфатаза В, гистаминаза, лейкотриены, катионные белки гранул эозинофилов.
Целый ряд других клеток – моноциты, эозинофилы и тромбоциты – также имеют на своей поверхности рецепторы для фиксирования реагинов, но их концентрация меньше, чем на тучных клетках и они низкоаффинные, с ними тоже взаимодействуют аллергены, в результате чего клетки высвобождают целый ряд различных медиаторов, обладающих провоспалительной активностью – это дополнительный путь.
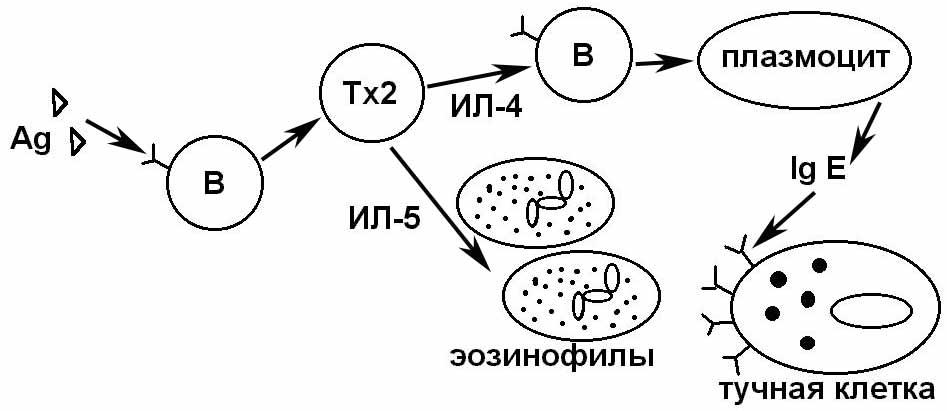
Рис. 12.8. Схема аллергической реакции реагинового типа.
Классический путь приводит к появлению немедленных реакций, развивающихся в первые полчаса. Дополнительный путь приводит к развитию так называемой поздней (или отсроченной) фазы аллергической реакции немедленного типа, развивающейся через 4-8 часов. Циркулирующий Ig Е метаболизируется довольно быстро, его период полужизни 2,4 суток. Фиксированный на клетках Ig Е сохраняется намного дольше около 28 суток.
Организм идёт по пути реагиновой гиперчувствительности при введении очень малых доз аллергена.
На малые дозы антигена макрофаги не реагируют и он представляется В–лимфоцитами Тн2, который увеличивает секрецию ИЛ4, а он стимулирует образование этих же клеток (В-клеток), идущих по пути образования Ig Е.
Атопические заболевания: атопическая форма бронхиальной астмы, поллиноз, атопический дерматит, и соответствующие формы крапивницы, пищевой и лекарственной аллергии, а также ряд гельминтозов, - в процессе эволюции Ig Е-механизм выработался для потивопаразитной защиты.
Процесс секреции медиаторов требует энергетического обеспечения. В этом процессе определенную роль играют циклические нуклеотиды клеток: цАМФ и цГМФ. От их соотношения зависит освобождение медиаторов.
Все стимулы, приводящие к накоплению цАМФ, тормозят высвобождение гистамина и некоторых других медиаторов. Поэтому эффективны адреналин и -адреномиметики. При анафилактическом шоке необходимо вводить адреналин. Ацетилхолин цГМФ выброс медиаторов. Поэтому эффективны холинолитики.
II. Цитотоксический тип, так его называют потому, что образовавшиеся к антигенам клеток антитела соединяются с клетками и вызывают их повреждение или даже лизис.
Повреждение может быть вызвано тремя путями:
За счет активации комплемента – комплемент–опосредованная цитотоксичность. С5–С9 комплемента образуют перфорины в клетках и содержимое клетки вытекает.
За счет активации фагоцитоза клеток, покрытых антителами (опсонизация).
Через активацию антител-зависимой клеточной цитотоксичности.
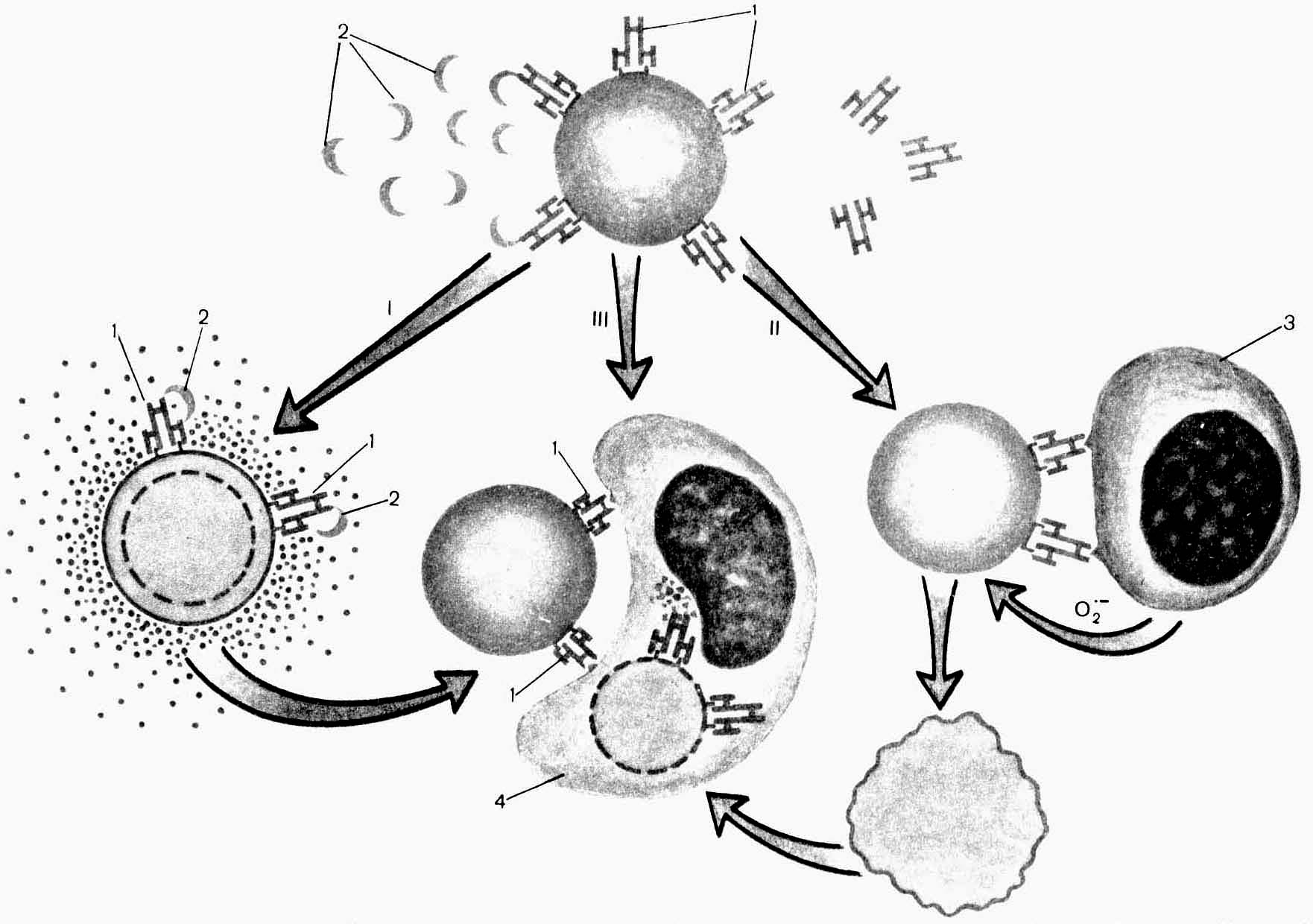
Рис. 12.9. Виды цитотоксичности. I – Комплемент-опосредованная цитотоксичность (С5–С9 комплемента образуют перфорины в клетках и содержимое клетки вытекает). II - Активация фагоцитоза клеток, покрытых антителами (опсонизация). III - Активация антител-зависимой клеточной цитотоксичности (К-клетки, имеющие рецепторы к Fc-фрагменту Ig, распознают опсонизированные Ag и "убивают" их с помощью активных форм кислорода).
Для того чтобы включился этот механизм, клетки должны приобрести аутоаллергенные свойства. Это могут быть химические вещества, лекарственные и бактериальные энзимы, вирусы, лизосомальные ферменты фагоцитов могут повреждать клетки, придавая им антигенные свойства. Поэтому многие паразитарные, бактериальные и особенно вирусные инфекционные заболевания сопровождаются образованием аутоантител к различным клеткам тканей и развитием гемолитической анемии, тромбоцито-лейкопении. При гепатите В, антитела, а не сам вирус вызывают гибель клеток печени, на поверхности которой и имеются антигены вируса гепатита В. Этот же механизм включается при попадании в организм аллоантигенов, например, при переливании крови, несовместимой по групповым или резус антигенам.
В зависимости от характера антител (их класс, подкласс) и их количества включаются различные пути повреждения. Антитела класса М, G1, G3, меньше G2 обладают способностью активировать комплемент. Другие антитела обладают опсонизирующими свойствами и обычно не фиксируют или слабо фиксируют комплемент. В третьем случае к Fс–фрагменту антитела присоединяются К–клетки, осуществляющие повреждающее действие на клетки мишени.
III. Повреждение иммунными комплексами (ИК). В норме в организме человека постоянно образуются иммунные комплексы и разрушаются ретикуло–эндотелиальной и фагоцитарной системой. Это один из способов элиминации антигена. AG в этих случаях присутствует в растворимой форме (бактериальные, вирусные, грибковые, лекарственные препараты, пищевые вещества). Образующиеся антитела относятся главным образом к классам Ig G, М.
В определенных условиях такой ИК может откладываться в тканях, чему способствует
повышение проницаемости сосудистой стенки;
образование комплекса в небольшом избытке Ag, что делает его не фагоцитабельным;
снижение активности фагоцитирующих клеток, что ведет к угнетению процесса очищения организма от ИК,
большое поступление или образование в организме антигенов.
ИК могут образовывать место в тканях или кровотоке, в зависимости от пути поступления или места образования.
ИК взаимодействует с комплементом. Образующие его активные фрагменты обладают хемотаксической активности, стимулирует активность нейтрофилов, повышают проницаемость сосудов и способствуют развитию воспаления. Нейтрофилы фагоцитируют ИК и при этом регенерируют супероксидные радикалы, выделяют лизисомные ферменты. Активирует калликреин–кининовая система, система свертывания и т. д. В результате происходит повреждение тканей – реакция на повреждение воспаление.
3 тип аллергических реакций является ведущим в развитии сывороточной болезни, некоторых случаев лекарственной и пищевой аллергии, ряда аутоаллергических заболеваний (ревматоидный артрид, системная красная волчанка).
35. – Этиология опухолей. Теории канцерогенеза. Канцерогены, проканцерогены, коканцерогены, синканцерогены.
Условия возникновения опухолей (факторы риска)
Дурные привычки
Табакокурение (около 90% случаев рака легкого - наиболее частой формы рака у мужчин, 5% общей медицинской смертности); смертность от рака легкого прямо пропорциональна числу выкуриваемых в день сигарет: у людей, выкуривающих 16-25 сигарет в день, риск заболеть раком легкого в 30 раз выше, чем у некурящих.
|
|
А |
Б |

Рис. 13.4. Рак легкого (А – макропрепарат, Б – электронная микрофотография).
Чрезмерное употребление алкоголя (опухоли желудка, полости рта, глотки, печени), вместе с курением дает кумулятивный эффект (синканцерогенез).
Факторы питания: диета, богатая жирами животного происхождения и копчеными продуктами, с высоким содержанием соли и низким содержанием грубых растительных волокон, с высокой концентрацией нитратов и пестицидов способствует новообразованиям; напротив, диета, включающая овощи, фрукты и витамины (С, А, -каротин), оказывает защитный эффект.
Многочисленные случайные половые связи (промискуитет), повышающие вероятность опухолей с вирусной этиологией (например, инфицирование вирусом папилломы человека при раке шейки матки).
Чрезмерный загар (высокий риск возникновения меланом, особенно у лиц с пониженной способностью к образованию меланина - у голубоглазых блондинов).
Условия труда - около 100 веществ, с которыми человек сталкивается в своей трудовой деятельности, являются предположительно канцерогенными (анилиновое производство, производство асбеста, асфальта и т.д.).
Классическим примером профессионального рака является описанный в 1897 г. рак мошонки у трубочистов в Лондоне. Рак рассматривался как профессиональное заболевание, которым страдали трубочисты в возрасте 30-35 лет (до сих пор остается непонятным вопрос о локализации опухоли именно в мошонке). Трубочисты, очищая дымоходы, втирали себе в кожу сажу и через 10-15 лет заболевали раком кожи. Объяснение механизмов развития этой формы рака послужило началом новой эры в исследовании опухолевого процесса. Было выяснено два основных фактора вызывающих развитие рака - постоянное раздражение, повреждение и действие определенных веществ (сажи), которые были названы канцерогенами. Эта модель заболевания была воспроизведена японскими учеными, которые в течение года втирали в ухо кролика сажу и получили сначала доброкачественную (папиллому), а затем и злокачественную опухоль.
Экологические факторы - к их числу относятся загрязнение водоемов и атмосферы промышленными отходами, содержащими канцерогены и радиоактивные соединения; загрязнение пищевых продуктов пестицидами и нитратами; строительство промышленных зданий и жилых помещений с использованием материалов, содержащих канцерогенные вещества (радон, фенолы и т.п.).
Канцерогенез (бластомогенез) – это процесс возникновения опухолей.
Канцерогены (канцерогенные или бластомогенные факторы) – это все факторы, вызывающие опухоли.
Коканцерогены – это агенты, усиливающие действие канцерогенов, но сами не вызывающие опухоли или вызывающие их крайне редко (например, лечение глюкокортикоидами и иммунодепрессантами).
Синканцерогены – это совместно действующие канцерогены с возможным потенцированием эффектов.
У курильщиков способствуют развитию рака легких следующие факторы - высокая температура, которая создается при курении, хронические бронхиты - вызывающие активную пролиферацию, и в табаке содержатся метилхолантрены - сильные канцерогены.
У моряков профессиональным заболеванием является рак кожи лица (воздействие ветра, воды, ультрафиолетового излучения солнца).
Проканцерогены (преканцерогены) – соединения, которые могут превращаться в канцерогены в организме (например, нитриты и нитраты под влиянием HCl желудка превращаются в нитрозамины; полициклические ароматические углеводороды эндогенно превращаются в эпоксиды).
Непосредственные причины опухолей (теории канцерогенеза)
Вирусно-генетическая теория Зильбера объясняет развитие опухолей под воздействием биологических канцерогенов – вирусов.
На сегодняшний день существует три «доказанных» онковируса:
ДНК-вирус Эпштейн-Барра (вызывает лимфому Беркитта и рак носоглотки или назофарингеальный рак);
вирус паппиломы человека (вызывает рак шейки матки);
ретровирус Human-N-leukemia virus, HTLV-1 (вызывает Т-клеточный лимфолейкоз).
ретровирус Human-N-leukemia virus, HTLV-3 (вызывает предположительно саркому Капоши);
вирус гепатита В (вызывает предположительно рак печени).
ДНК-вирусы могут сразу внедряться в геном, начиная разрушительную работу, инициируя некотролируемое деление и др.
РНК-вирусы (онкорновирусы), чаще всего это ретровирусы, имеющие фермент обратная транскриптаза (синтез молекул ДНК на информационной РНК вируса).
Физико-химическая теория Вирхова.
Физические канцерогены
Высокая (реже низкая) температура.
Повторные ожоги могут вызвать так называемый «ожоговый» бытовой рак «кангри» (в северных областях Индии при использовании для согревания глиняных горшков с горячими углями, укрепляемых под одеждой на животе).
У северных народов наблюдается более высокая частота заболеваемости раком пищевода в связи с употреблением очень горячей пищи - горячей рыбы.
Механическое раздражение (например, зубными протезами).
Ионизирующая радиация.
К ионизирующей радиации относятся рентгеновское излучение(квантовое электромагнитное излучение), α-излучение (поток положительно заряженных ядер гелия, обладает высокой ионизирующей, но малой проникающей способностью), -излучение (поток электронов со сравнительно высокой проникающей, но низкой ионизирующей способностью), γ-излучение (квантовое электромагнитное излучение с длиной волны, меньшей, чем у рентгеновского излучения). Поток нейтронов (электрически нейтральных частиц) обладает высокой проникающей способностью и, сталкиваясь с атомами различных веществ, вызывает вторичное α-, -, γ-излучение.
У людей рентгеновские лучи могут быть причиной так называемого профессиовального («рентгеновский» рак кожи и лейкоз у рентгенологов) и ятрогенного рака (опухоли, возникающие после врачебного лечения).
Солнечная и ультрафиолетовая радиация. Длительное воздействие УФ спектра солнечных лучей является основным индуктором меланом на открытых участках кожи (голова, шея, руки).
Химические канцерогены
Органические канцерогены
полициклические углеводороды (ПАУ) - наиболее важные из них, «сильные» канцерогены, бенз(а)пирен (содержится в сигаретах, в почве, в выбросах вулканов) и диметилбенз(а)антрацен; становятся конечными канцерогенами в организме, превращаясь в соответствующие эпоксиды;
ароматические амины и амиды - нафтиламин, бензидин вызывают профессиональный рак мочевого пузыря;
аминоазосоединения - ортоаминоазотолуол, диметиламиноазобензол;
нитрозосоединения - диметилнитрозамин, диэтилнитрозамин, этил- и диэтилнитрозомочевина; могут синтезироваться в организме из нитритов (нитратов) и вторичных аминов, содержащихся в пище; кроме того, вторичные амины могут образовываться в толстой кишке при участии бактериальной флоры, которая в то же время способна переводить нитраты в нитриты
афлатоксины - вещества, образуемые плесенью Aspergilus flavus, поражающей пищевые продукты (особенно арахис);
другие органические канцерогенные вещества, относящиеся к различным классам соединений - уретан, этионин, четыреххлористый углерод, хлорэтиламины, эпоксиды, лактоны, винилхлорид, пластмассы, липидные перекиси и др.
Неорганические канцерогены: хром, мышьяк (примесь его в сигаретах в 15 раз превышает его максимально допустимое количество), кобальт, никель (содержится в табачном дыме), бериллий, свинец, кадмий и др.
Асбест широко использовался как изоляционный и огнеупорный материал, найден почти во всех постройках, возведенных в США с 1940 по 1970 год. Самое большое индивидуальное поражение асбестом происходило у рабочих верфей во время второй мировой войны.
Лекарственные канцерогены
Рентгеноконтрастный препарат торотраст (включавший окись тория) применялся в 1930-1945 годах для диагностических целей при заболеваниях печени и селезенка и способствовал спустя 15–20 лет возникновению злокачественных опухолей. Отлагаясь в костях близ кроветворных тканей, природный торий-232 становится источником гораздо более опасных для организма изотопов - мезотория, тория-228, торона.
Противоопухолевые лекарства - некоторые лекарства, используемые для лечения опухолей (алкилирующие агенты, типа циклофосфамида, хлорамбуцила, бисульфана и тиотефа) воздействуют на синтез нуклеиновых кислот и в опухолевых клетках, и в нормальных клетках и могут вызывать онкогенные мутации (например, лейкемия - наиболее частое неопластическое осложнение химиотерапии рака).
Эстрогены – вызывают гиперплазию эндометрия, которая сопровождается сначала цитологической дисплазией, переходящей затем в рак эндометрия.
Предполагаемый механизм образования ДНК-повреждающих реактивных метаболитов в процессе обмена эстрогенов (J.Liehr, 1990) заключается в том, что благодаря ферментативным реакциям в тканях-мишенях (молочная железа, эндометрий и др.) из классических эстрогенов типа эстрадиола или эстрона образуются катехолэстрогены, в ходе метаболического восстановительного цикла возникают свободные радикалы, в частности семихиноны и их производные, которые в свою очередь индуцируют образование других свободных радикалов, включая супероксидный анион и перекиси липидов, которые могут повреждать белки и ДНК.
Диэтилстилбэстрол – синтетический эстроген, использовался в высоких дозах с 1950 по 1960 год для лечения угрожающего выкидыша. У детей, которые внутриутробно были подвержены влиянию диэтилстилбэстрола, было определено значительное увеличение заболеваемости светлоклеточной аденокарциномой, которая является редким раком влагалища и развивается у молодых женщин между 15 и 30 годами.
Стероидные гормоны – использование оральных контрацептивов и анаболических стероидов иногда связывают с возникновением доброкачественных печеночноклеточных аденом, описано несколько случаев возникновения печеночноклеточного рака.
Препараты различных групп, вызывающие гинекомастию (некоторые с известным стероидным эффектом, механизм действия других неясен), которая является предраковым заболеванием (рак молочной железы у мужчин): изониазид, ранитидин, метронидазол, метилдофа, эналаприл, амиодарон.
Полиэтиологическая теория объединяет две выше упомянутые теории.
Генотоксическая (современная) теория
Превращение нормальной клетки в опухолевую является результатом стойких изменений в геноме клетки. Т.е. все канцерогены генотоксичны, поскольку вызывают повреждение ДНК. С открытием молекулярных механизмов канцерогенеза появился новый взгляд на этиологию – генотоксическая теория. Вирусы, физические факторы и химические вещества - это только виды канцерогенов.
36. – Патогенез опухолей. Стадии опухолевого роста и их механизмы. Виды атипизмов. Понятие и значение онкомаркеров. Стадии и механизмы метастазирования.
Молекулярные механизмы опухолевого роста
У делящейся клетки с поврежденной ДНК есть выбор: либо приостановка деления до полной репарации повреждений, либо самоуничтожение путем апоптоза. Гибель одной клетки не может иметь никаких отрицательных последствий, а ее сохранение таит смертельную угрозу возникновения клона дефектных (потенциально опухолевых) клеток. Можно предполагать, что опухолевый рост возможен лишь потому, что дефектные клетки способны каким-то образом избегать апоптоза.
Существует несколько механизмов, повреждение которых может способствовать опухолевому росту:
промитогенные факторы (протоонкогены и протоонкобелки);
антимитогенные факторы (супрессоры) активностью;
механизмы защиты (репарация ДНК и апоптоз).
На периферии клетки существуют рецепторы ростовых факторов, воспринимаемые рецепторами внешние сигналы в виде каскадов реакций фосфорилирования передаются внутрь клетки. Перенос митогенного сигнала от периферии клетки к ее генетическому аппарату осуществляется в виде каскада реакций фосфорилирования посредством протеинкиназ (ферментов, фосфорилирующих белки). Волна митогенной импульсации в упрощенном виде сводится к передаче фосфатной группы, наподобие эстафетной палочки, от одной протеинкиназы к другой. В конечном итоге она достигает ядерных регуляторных белков (транскрипционных факторов), активирует их (тоже посредством фосфорилирования) и тем самым индуцирует перепрограммирование генома. После обработки сигнала в клеточном ядре поток молекул мРНК индуцирует митотическую активность.
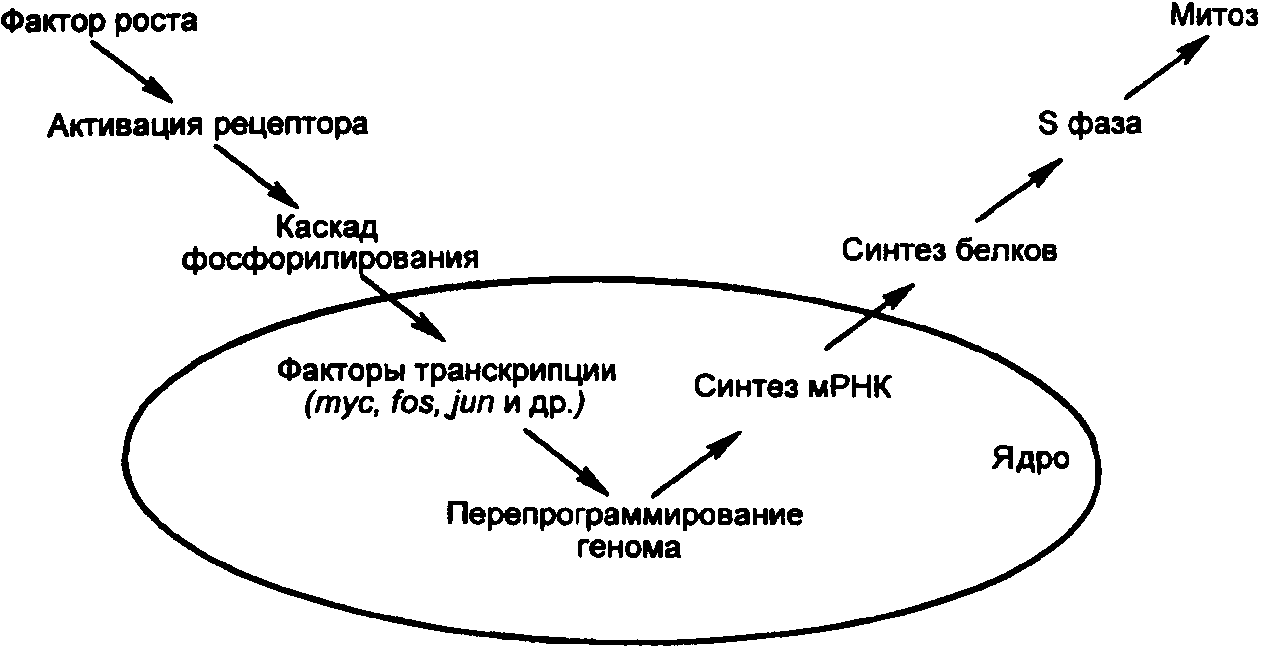
Рис. 13.5. «Рефлекторная дуга» митогенного сигнала.
Нерегулируемое размножение измененной клетки можно представить, как возникновение очага «застойного» возбуждения в том или ином звене пути переноса митогенного сигнала. Повреждение гена и как следствие - структурный дефект какого-либо из сигнальных белков, способный зафиксировать его в постоянно активном состоянии (т.е. сделать независимым от «вышестоящих» регуляторных инстанций), один из главных механизмов канцерогенеза. Нормальные гены, участвующие в переносе митогенного сигнала и потенциально способные на такое превращение, называются протоонкогенами.
Протоонкогены являются акселераторами клеточного деления и в случае превращения в онкогены проявляют себя как доминантный признак.
В нормальных клетках присутствуют факторы, тормозящие клеточное деление, блокирующие каждый своим способом разные этапы митогенной стимуляции, это гены-супрессоры. Одним из таких генов является ген р53, продукт этого гена является регулятором транскрипции, его называют «главным хранителем генома». Деятельность р53 проявляется в момент вхождения клетки в фазу синтеза ДНК (граница фаз G1/S, когда проверяется целостность ДНК), в случае неудовлетворительной целостности ДНК запускается апоптоз.
Полная трансформация клетки, таким образом, является следствием нескольких генетических событий: активации онкогена(ов) и инактивации гена(ов), осуществляющих супрессорные функции.
Два типа генов управляют размножением клеток: протоонкогены, которые играют роль акселераторов деления, и гены-супрессоры, выполняющие функцию тормозов. Заклиньте акселератор или уберите тормоза - и клетка, будто спущенная с цепи, начнет безостановочно делиться (J.M. Bishop, лауреат Нобелевской премии 1989 г.).
Стадии канцерогенеза
Индукция (инициация) заключается в мутации одного из генов, регулирующих клеточное размножение (протоонкоген превращается в онкоген) → клетка становится потенциально способной к неограниченному делению; инициирующими факторами являются различные канцерогены.
Промоция (ускорение) - стимуляция клеточного деления промоторами, благодаря которой создается критическая масса инициированных клеток Промоторы – это химические вещества, не вызывающие повреждения ДНК, не являющиеся канцерогенами. Свою деятельность начинают онкогены → синтезируются онкобелки → количество инициированных клеток увеличивается.
Прогрессия - наряду с увеличением массы опухоли она постоянно приобретает новые свойства, «озлокачествляется» - все большую автономность от регулирующих воздействий организма, деструктивный рост, инвазивность, способность к образованию метастазов (обычно отсутствующую на ранних этапах) и, наконец, приспособляемость к меняющимся условиям.
Опухоль представляет собой потомство (клон) одной первичной клетки, которая в результате многостадийного процесса приобрела способность нерегулируемого роста. Первичная трансформированная клетка передает свои свойства только своим потомкам, т.е. «вертикально». При этом окружающие опухоль нормальные клетки в процесс перерождения не вовлекаются. Это представление получило название положения о клональном происхождении опухоли.
Клональная гетерогенность опухоли развивается из-за генетической нестабильности опухолевой клетки. Это приводит к появлению новых клонов, различающихся генотипически и фенотипически. В результате селекции отбираются и выживают самые злокачественные клоны. После химиотерапии остается всего 0,1% клеток опухоли, но так как клеточный цикл равен 24 часам, то опухоль может восстанавливаться через 10 суток и быть резистентной к прежней химиотерапии.
Рис. 13.6. Стадии развития опухоли на примере формирования рака легкого.
Свойства опухолевого роста. Атипизмы. Влияние опухоли на организм.
Атипизм (от а + греч. typicos - образцовый, типичный) - совокупность признаков, отличающих опухолевую ткань от нормальной, и составляющих биологические особенности опухолевого роста.
Анаплазия или катаплазия (от ana - обратное, противоположное, kata - вниз + греч. plasis - формирование) - изменение структуры и биологических свойств опухоли, делающее их похожими на недифференцированные ткани.
Термин введен ввиду определенного формального сходства опухолевых клеток с эмбриональными (интенсивное размножение, усиленный анаэробный гликолиз). При этом опухолевые клетки принципиально отличаются от эмбриональных. Они не созревают, способны к миграции и инвазивному росту в окружающие соседние ткани с разрушением их и т.д.
Биологические особенности, характерные для доброкачественных и злокачественных опухолей
Атипизм размножения - бесконтрольное деление опухолевых клеток.
нерегулируемое размножение клеток (например, ослабление свойств опухолевых клеток тормозить митотический цикл и передвижение при контакте друг с другом, т.е. отсутствие контактного торможения);
утрата верхнего «лимита» числа делений клетки (так называемого лимита Хайфлика): нормальные клетки делятся до определенного максимального предела (у млекопитающих в условиях клеточной культуры до 30-50 делений), после чего они погибают, а опухолевые клетки приобретают способность к бесконечному делению (иммортализация - «бессмертие» данного вида клеток).
Атипизм регуляции роста и дифференцировки (дедифференцировка) - частичное или полное подавление процесса созревания клеток.
Опухоль приобретает сходство с эмбриональными клетками, (мало митохондрий, рецепторов, особые белки-фетопротеины), при этом созревание остановлено, нет специализации, обучения клеток.
Этот атипизм резко выражен у злокачественных опухолей и слабо - у доброкачественных. Причинами его являются утрата опухолью факторов, стимулирующих дифференцировку ее клеток, или пониженная чувствительность клеток к ним.
Процессы роста, дифференцировки и деления в норме находятся под контролем центральной эндокринной регуляции, которая осуществляется соматотропным гормоном, гормонами щитовидной железы, инсулином.
Кроме этих общих факторов, в каждой ткани существуют свои факторы роста и дифференцировки (фактор роста эпидермиса, тромбоцитарный фактор, интерлейкины).
Индукция роста и дифференцировки начинается с взаимодействия фактора роста с рецептором фактора роста на клеточной мембране (в опухолевой клетке этот этап может быть нарушен). На следующем этапе образуются вторичные посредники - циклические аденозин- и гуанозинмонофосфат, причем для нормального роста и дифференцировки характерно преобладание цАМФ. Образование цГМФ сочетается с усилением пролиферации. В опухолевых клетках это типичный признак.
Биохимический атипизм новообразований включает:
интенсивный синтез онкобелков («опухолеродных» или «опухолевых» белков).
Эти белки обусловливают появление у клеток обязательных опухолевых особенностей (бесконтрольное деление, утрата лимита деления, иммортализация и др.). Синтез онкобелков программируется активными клеточными онкогенами. Активные онкогены выявляются только в опухолевых клетках, протоонкогены - во всех нормальных клетках.
уменьшение синтеза и содержания гистонов (белков-супрессоров синтеза ДНК).
Дефицит гистонов способствует активации синтеза ДНК- и РНК-матриц, что в свою очередь приводит соответственно к удвоению генов, хромосом, белковой массы и к делению клеток.
образование несвойственных здоровым клеткам белков (например, α-фетопротеина) и других веществ, называемых онкомаркерами (позволяют обнаружить рецидив, или метастазы опухоли на 3,5 месяца раньше появления клиники).
α-фетопротеин (АФП) синтезируется в норме в антенатальном периоде гепатоцитами плода (от лат. fetus - плод), но почти не образуется постнатальными, «зрелыми» гепатоцитами.
изменение способа ресинтеза АТФ заключается в увеличении доли АТФ, образуемой в ходе гликолиза (анаэробного и аэробного) и уменьшения, соответственно, доли АТФ, ресинтезируемой в процессе тканевого дыхания (аэробного окисления).
В нормальных клетках и тканях в анаэробных условиях усиливается гликолиз. В присутствии кислорода он ингибируется (положительный эффект Пастера). Напротив, в опухолевых клетках интенсивный анаэробный гликолиз при смене анаэробных условий на аэробные не снижается, а сохраняется (отрицательный эффект Пастера).
Усиление гликолиза в опухолевых клетках обусловливает их высокую выживаемость в условиях гипоксии.
Преобладание гликолиза приводит к повышению концентрации молочной кислоты в клетках опухоли, характерен ацидоз, приводящий к нарушению жизнедеятельности самой клетки (зона некроза расположена обычно в центре опухоли).
феномен субстратных «ловушек» заключается в усиленном захвате и использовании субстратов для энергообразования (глюкозы), для построения цитоплазмы (аминокислот - отсюда «ловушка азота»), мембран клеток (холестерина), для защиты от свободных радикалов и стабилизации мембран (например, антиоксиданта - токоферола).
Эта особенность повышает выживаемость опухолевых клеток при контакте их с нормальными клетками в условиях инвазивного роста и метаста-зирования.
снижение содержания в клетках опухолей цАМФ, оказывающего, как правило, тормозное влияние на их деление и увеличение цГМФ, стимулирующего пролиферацию клеток.
Физико-химический атипизм проявляется увеличением содержания в опухолевых клетках воды, ионов калия и уменьшением в них кальция и магния.
увеличение содержания воды облегчает диффузию субстратов метаболизма внутрь клеток и его продуктов наружу.
снижение содержания Са2+ уменьшает межклеточную адгезию, а это в свою очередь облегчает «отшнуровывание» клеток от ткани опухоли и движение их в окружающие нормальные ткани при инвазивном росте.
увеличение содержания К+ препятствует в определенной мере развитию внутриклеточного ацидоза в связи с усилением гликолиза и накоплением молочной кислоты.
повышается величина отрицательного заряда поверхности опухолевых клеток, что способствует увеличению их взаимоотталкивания и проникновению по межклеточным щелям в нормальные ткани. Увеличение отрицательного заряда поверхности клеток происходит вследствие накопления на ней анионов нейраминовой кислоты.
повышается электропроводность и снижается вязкость клеточных коллоидов.
опухолевые клетки излучают в большом количестве митогенетические лучи Гурвича (ультрафиолетовые лучи с длиной волны 190-325 нм, способные стимулировать деление соседних клеток).
Функциональный атипизм проявляется нарушением функций клеток:
снижение секреции желудочного сока при раке желудка, образования желчи при раке печени и т.д.
неадекватное, нецелесообразное усиление функций, например повышение синтеза инсулина инсулиномой - опухолью из клеток островков Лангерганса поджелудочной железы вызывает гипогликемическое состояние, а в ряде случаев - гипогликемическую кому.
«извращение» функций, например синтез опухолевыми клетками при раке молочной железы гормона щитовидной железы кальцитонина; синтез клетками при раке легких некоторых гормонов передней доли гипофиза - АДГ, АКТГ и др.
Антигенный атипизм состоит в разнонаправленных изменениях антигенного состава опухолевых клеток (антигенном упрощении или появлении новых антигенов).
антигенное упрощение - утрата опухолевыми клетками антигенов, имеющихся в исходных нормальных клетках (например, утрата раковыми гепатоцитами органоспецифического печеночного антигена, h-антигена).
появление новых антигенов, отсутствовавших в нормальных (например, эмбрионального антигена α-фетопротеина в раковых гепатоцитах).
Утрата клетками новообразований органоспецифического антигена и появление в них эмбриональных антигенов (к которым не образуются антитела, так как они воспринимаются иммунной системой как свои) способствуют антигенной «маскировке» опухолевых клеток и «неузнаваемости» их иммунной системой.
Морфологический атипизм делят на тканевый и клеточный.
Тканевый атипизм заключается в нарушении нормального соотношения тканевых структур.
Клеточный атипизм проявляется полиморфизмом - разной формой и размерами клеток (клеточный полиморфизм) и ядер (ядерный полиморфизм); увеличением ядерно-цитоплазматического отношения; гиперхромией ядер; изменением числа, формы и размеров хромосом (хромосомные аберрации); увеличением количества свободнолежащих в цитоплазме рибосом, участвующих в синтезе белков, увеличением размеров и числа ядрышек в ядрах, увеличением числа митоза, появлением различных по величине и форме митохондрий.
Атипизм «взаимодействия» клеток опухоли с организмом.
Опухоль – «ловушка» питательных веществ, таких как глюкоза, азот, витамины развитие гипогликемии, анемии.
Изменение иммунного надзора (см. ниже).
Опухоль - источник биологически активных веществ: ростовые и ангиогенные факторы, эктопические гормоны (АКТГ при раке легкого), гиперпродукция или угнетение синтеза гормонов при опухолях эндокринных желез.
Паранеопластический синдром – проявление генерализованного воздействия опухоли на организм. Его формы разнообразны: состояние иммунодепрессии, гиперкоагуляция, сердечно-сосудистая недостаточность, мышечная дистрофия, пониженная толерантность к глюкозе, острая гипогликемия при опухолях больших размеров и др.
Одним из проявлений паранеопластического синдрома является раковая кахексия, которая возникает в периоде, близком к терминальному. Она характеризуется потерей массы тела в основном из-за усиленного распада белков скелетных мышц (частично миокарда), а также истощения жировых депо.
В развитие раковой кахексии вносят вклад ряд явлений, развивающихся в организме опухоленосителя:
нарушение нервно-эндокринной регуляции обмена веществ;
усиление образования АТФ за счет гликолиза, что повышает расход субстратов энергообразования;
ингибирование липопротеинлипазы, катализирующей накопление липидов в организме;
снижение синтеза РНК, обеспечивающих синтез белков и дифференцировку адипоцитов;
образование фактора некроза опухолей, он же - кахектин - цитотоксический полипептидный гормон, известный также как TNF (tumor necrosis factor — фактор некроза опухолей). Он секретируется макрофагами и опосредует, воспалительные реакции. Практически все клетки организма обладают рецепторами к этому гормону, эффекты которого поэтому могут быть весьма многообразными: шоковое состояние, падение артериального давления, расстройства липидного и углеводного обмена, метаболический ацидоз, активация нейтрофилов вплоть до гибели организма, состояния анорексии и истощения организма.
снижение синтеза каталазы накопление избытка продуктов свободно-радикального и перекисного окисления;
сопутствующие опухоли осложнения: боль, кровотечение, нарушение функций гастроинтестинальной системы; феномен улавливания опухолью субстратов из крови.
Кахексия может наблюдаться не только при злокачественных, но и при некоторых доброкачественных опухолях при их определенной локализации: в желудочно-кишечном тракте (вследствие развития непроходимости или резкого нарушения секреторной, моторной и всасывательной функций); в головном мозге, в области трофических центров (вследствие нарушения нервно-гормональной регуляции обмена веществ и энергии).
Биологические особенности, характерные для злокачественных опухолей
Инфильтративный (или инвазивный) рост заключается в проникновении клеток опухоли в окружающие нормальные ткани, сочетается с деструкцией этих тканей. Этому способствуют:
приобретение клетками способности к отделению от опухолевого узла и к активному перемещению.
образование опухолевыми клетками белковых веществ - «канцероагрессинов», проникающих в окружающие нормальные ткани и стимулирующих хемотаксис и благодаря этому - инвазию в них опухолевых клеток.
уменьшение сил клеточной адгезии - у опухолевых клеток уменьшается площадь поверхности соприкосновения, уменьшается количество нексусов - контактов, обеспечивающих адгезивность клеточных мембран, меняется состав мембранных гликопротеидов, что облегчает отделение опухолевых клеток и их последующее движение.
уменьшение контактного торможения.
Ангионеогенез – образование новых сосудов в олухоли, имеет исключительное значение, поскольку без него рост опухоли ограничивается 1-2 мм.
Стимулирует процесс новообразования сосудов белок ангиогенин (кроме того так же ФНО, ИЛ8 и др.). Сосуды содержат лишь базальную мембрану и эндотелий, мышечной оболочки нет, поэтому сосуды не могут менять просвет. При отставании васкуляризации в центре опухоли развивается некроз.
Тормозят васкуляризацию ангиостатины (α- и -интерфероны, гепариназа). Таким образом, опухоль сама регулирует свой ангионеогенез. Иногда хирургическое удаление опухоли провоцирует ангионеогенез.
Метастазирование (от греч. metastasis — перемена места, перемещение, перенос) - перемещение опухолевых клеток из первичной опухоли в органы и ткани, расположенные на расстоянии, и образование в них новых, вторичных, опухолевых узлов той же гистологической структуры.
Рис. 13.7. Метастазы остеогенной саркомы в паренхиму легких.
Различают следующие пути метастазирования опухолевых клеток:
лимфогенный - перенос клеток лимфой по лимфатическим сосудам;
гематогенный - транспорт их кровью по кровеносным сосудам;
гематолимфогенный - перенос и лимфогенным и гематогенным путем;
«полостной» - перенос опухолевых клеток жидкостями в полостях тела, например цереброспинальной;
имплантационный - прямой переход опухолевых клеток с поверхности опухоли на поверхность органа или ткани, с которыми она контактирует (например, имплантация опухолевых клеток рака верхней тубы на нижнюю).
Рис. 13.8. Этапы метастазирования.
В развитии лимфогенных, гематогенных и гематолимфогенных матастазов различают три стадии:
Стадия инвазии - проникновение опухолевых клеток через стенку сосудов в их просвет.
Стадия клеточной эмболии - перенос током лимфы или крови, проникших в просвет сосудов опухолевых клеток, остановка их в просвете микрососудов с последующим образованием на их поверхности нитей фибрина, что ведет к превращению клеточного эмбола в клеточный тромбоэмбол, прикрепляющийся к эндотелию.
Стадия проникновения опухолевых клеток из клеточного тромбоэмбола через стенку сосудов в окружающие нормальные ткани, размножение их с образованием новых опухолевых узлов.
Рецидивирование. Причинами рецидивов являются:
неполное удаление опухолевых клеток, чему способствует инфильтративный рост новообразования;
имплантация опухолевых клеток в окружающую нормальную ткань при травматично выполненной операции с нарушением правил абластики;
проникновение нуклеиновых кислот (ДНК онкогенов) в клетки окружающих нормальных тканей;
иммунодепрессия, возникающая в части случаев после операции.
Опухолевые маркеры. Механизмы изменения иммунного надзора при опухолях.
Опухолевые маркеры - соединения, обнаруживаемые в биологических жидкостях онкологических больных и синтезируемые либо собственно раковыми клетками, либо клетками нормальных тканей в ответ на инвазию опухоли.
Маркерами опухоли могут быть различные белки (ферменты, гормоны, антигены - внутриклеточные или ассоциированные с поверхностными мембранами) и метаболиты, концентрация которых в среде коррелирует с массой опухоли, с ее пролиферативной активностью и иногда со степенью злокачественности.
Маркёры, продуцируемые опухолью
Онкофетальные протеины
Раково-эмбриональный антиген (Carcinoembryonic antigen, cEA) - продукция повышается при раке толстой кишки, поджелудочной железы, молочной железы, легкого.
α-фетопротеин (Alpha fetoprotein, AFP) - продукция повышается при гепатоцелюлярном раке.
Гормоны
Хорионический гонадотропин (Horionic gonadotropin, HCG).
Аденокортикотропный гормон (Adrenocorticotropic hormone, ACTH) – продукция может повышаться при раке легкого.
Другие
Иммуноглобулины (например, белок Бенс-Джонса).
Простатический специфический антиген (Prostate-specific antigen, PSA).
Маркёры, ассоциированные с опухолью
Белки острой фазы воспаления (церрулоплазмин, гаптоглобин, α2-глобулины, С-реактивный белок) – повышение продукции связано со стимуляцией ростовыми факторами, секретируемыми опухолями, или вторичным воспалением.
Нормальные энзимы в высоких концентрациях (лактатдегидрогеназа (ЛДГ), креатинфосфокиназа (КФК)).
Гормоны
Аденокортикотропный гормон (Adrenocorticotropic hormone, ACTH).
37. – Механизмы зашиты организма от опухолей. Механизмы ускользания опухолей от иммунного контроля. Методы диагностики опухолевого роста. Профилактика опухолевого роста.
Противоопухолевый иммунитет
Естественный иммунитет.
Макрофаги и NK-клетки, особенно активированные, могут сдерживать рост опухоли (цитостаз) или разрушать её (цитолиз). При интенсивной стимуляции макрофаги выделяют фактор некроза опухоли (ФНО), который разрушает некоторые опухоли, но вызывает побочные эффекты - потерю массы тела и др. ФНО действует вместе с интерфероном. γ-ИФ необходим для активации макрофагов и NК-клеток.
NК-клеток (естественные или натуральные киллеры) весьма эффективны в противоопухолевой защите, поскольку распознают чужеродные клетки по отсутствию антигенов главного комплекса гистосовместимости (но NK-клеток мало, менее 1% из всей массы лимфоцитов).
Приобретенный (адаптивный) иммунитет.
Антитела эффективнее действуют против свободных клеток (лейкозы, метастазируюшие опухоли), чем против солидных опухолей. Антитела могут содействовать росту опухоли (эффект усиления), возможно, за счёт формирования комплексов с растворимыми антигенами и блокады опосредованной Т-клетками цитотоксичности. Антительный ответ более эффективен, чем активированные макрофаги, для защиты от химических канцерогенов.
Клеточный иммунитет. Т-хелперы активируют макрофаги и выделяют ФНО. Некоторые опухолевые клетки утрачивают в процессе формирования многие нормальные антигены, в том числе молекулы ГКГС, и становятся недоступными для Т-киллеров.
Супрессия. Опухоли из В-клеток, инфицированных вирусом Эпштейн-Барра (лимфома Беркитта), возникают из-за того, что Т-киллеры, подавленные Т-супрессорами, не могут выполнять свои защитные функции.
Механизмы «ускользания» опухоли от иммунного контроля
интоксикация иммуннокомпетентных органов;
слишком интенсивное размножение;
фибриновая пленка, мешающая распознаванию опухолевых антигенов;
антигенное упрощение иммунокомпетентной системе нечего распознавать;
антигенный туман - синтез нефиксированных антигенов вокруг опухоли, на которые и «бросаются» специфические иммунокомпетентные клетки и антитела;
синтез собственных экранирующих антител-колпачков на чуждые антигены;
активация Т-супрессоров;
увеличение продукции глюкокортикоидов, угнетающих иммунитет, в ответ на гипогликемию.
Рис. 13.9. схема противоопухолевого иммунитета.
Принципы фармакологического торможения опухолевого роста.
Главное направление этой области фармакотерапии - препятствовать повышенной митотической активности опухолевой клетки.
С этой целью применяются цитостатики, или цитотоксичные препараты (допан, циклофосфан, тиотэф, миелосан). Поскольку данная группа препаратов не может действовать избирательно только на опухолевые клетки, в значительной степени страдают и те органы и ткани, в которых изначально велика интенсивность клеточного деления: эпителиальная (что может проявиться в виде плохого заживления ран после операций) и, особенно, кроветворная система (что может проявиться анемией и лейкопенией). Тем более, что данные симптомы возникают и вследствие интоксикации красного костного мозга продуктами жизнедеятельности опухолевых клеток, а также феноменом "ловушек" аминокислот и глюкозы, каковыми являются опухолевые клетки с их неестественно повышенным метаболизмом. Последнее требует коррекции прежде всего в виде стимуляторов эритро- и лейкопоэза.
Поскольку среди условий возникновения опухолей, а также неизбежных звеньев патогенеза присутствует ослабление иммунной системы, лечение опухолей включает назначение иммуностимуляторов в качестве этиотропной и патогенетической терапии. В последние годы предпринимаются успешные попытки лечения лейкокинами интерлейкинами (ИЛ2, α-интерферон).
БЦЖ (ослабленные туберкулёзные бациллы) применяется для лечения больных с меланомой, саркомой и другими опухолями. Главное воздействие направлено на активацию макрофагов и NK-клеток. Большое количество бактериальных и других иммуностимулирующих агентов используется в активации противоопухолевой защиты.
Пассивная иммунизация моноклональными антителами к опухолеассоциированным антигенам, конъюгированными с токсическими препаратами и др., могут усилить разрушение опухоли.
Из соображений возможности ятрогенного канцерогена, надо также с осторожностью (особенно при предопухолевых состояниях) применять лекарственные препараты, содержащие в составе тяжелые металлы и их аналоги, полициклические ароматические углеводороды (например, стероидные структуры), ароматические амины (например, производные анилина), нитриты и нитраты (возможность образования нитрозаминных групп). Вместе с тем, учитывая высокую интенсивность метаболизма в опухолевых клетках, делаются попытки заблокировать его путем назначения именно тяжелых металлов как блокаторов многих ферментов. Еще издавна делались попытки лечить опухоли препаратами ртути, в частности, сулемой HgCl2, но из-за высокой токсичности последней метод не получил распространения. Теперь этот подход переживает второе рождение. Менее токсичные органические препараты ртути более избирательно поражают опухолевые клетки благодаря идее "кондуктора" проводника, который интенсивно поглощается именно опухолевой клеткой и проводит за собой нужную часть молекулы противоопухолевого препарата.
Несмотря на то, что, как говорилось выше, многие стероиды могут служить по крайней мере коканцерогенами и синканцерогенами (особенно половые гормоны при предраковых состояниях в своих органах - мишенях), при гормон-зависимых опухолях половой сферы применяют блокаторы секреции половых гормонов за счет торможения секреции гонадотропных гормонов (леупролид, гозерелин) и блокаторы рецепторов к половым гормонам – антигормоны (антагонист эстрогенов тамоксифен или антагонист андрогенов флутамид). Оригинален метод лечения таких гормон-зависимых опухолей половыми гормонами противоположного пола, что основано на выключении секреции собственных половых гормонов по механизму обратной связи.
Некоторые опухолевые клетки (лимфобласты при остром лимфолейкозе) не могут синтезировать необходимый им метаболит (аспарагинат), поэтому для большего уменьшения поступления этой аминокислоты в опухолевые клетки применяют введения в организм фермента (L-аспарагиназа).
Среди других групп лекарственных препаратов, применяющихся при опухолях, следует отметить еще ряд адаптогенов растительного и животного происхождения, повышающих неспецифическую резистентность организма (апилак, чага, жень-шень), а также другие средства симптоматической терапии препараты для нормализации белкового и углеводного состава плазмы крови, при необходимости обезболивающие.
38. – Патология углеводного обмена. Гипогликемические состояния. Нарушения всасывания, синтеза, депонирования и расщепления углеводов.
Углеводы – это альдегидоспирты (кетоспирты) с одной карбонильной и несколькими гидроксильными группами, а также их производные.
В организме углеводы выполняют следующие функции:
Источник энергии (обеспечивают около 67% суточного потребления энергии).
Резерв энергии (в виде гликогена).
Пластическая функция (продукты обмена участвуют в синтезе липидов, аминокислот, мукополисахаридов тканей).
Защитная функция (углеводные компоненты иммуноглобулинов участвуют в иммунитете).
Источник углеводов в питании человека - преимущественно пища растительного происхождения. Суточная потребность в углеводах составляет 400-500 г.
Пищевые углеводы в основном представлены крахмалом, сахарозой (преобладают в пище взрослых людей) и лактозой (является основным углеводом в питании детей грудного возраста).
Переваривание углеводов начинается в тонком кишечнике. Кратковременное воздействие амилазы слюны на крахмал пищи существенной роли не играет, так как в просвете желудка кислая среда инактивирует этот фермент.
В тонком кишечнике крахмал под действием амилазы поджелудочной железы, выделяющейся в двенадцатиперстную кишку с панкреатическим соком, расщепляется до мальтозы и изомальтозы. Эти дисахариды, а также лактоза и сахароза, расщепляются специфическими ферментами (гликозидазами), продуцируемыми слюнными железами, поджелудочной железой и щеткообразным эпителием кишечника. Ферменты работают не в просвете кишечника, а на поверхности клеток (это т.н. пристеночное пищеварение). В результате образуются моносахариды (глюкоза, фруктоза, галактоза), которые затем транспортируются через плоский кишечный эпителий в воротную систему крови.
Всасывание моносахаридов из кишечника происходит с помощью специальных белков-переносчиков (транспортеров). Кроме того, глюкоза и галактоза транспортируются в энтероцит путем активного транспорта, зависимого от градиента концентрации ионов Na+. Он обеспечивает перенос глюкозы в энтероцит против ее градиента концентрации. Энергия, необходимая для этого активного транспорта, обеспечивается Nа+/К+-АТФазой. Это обеспечивает всасывание глюкозы и галактозы даже при низкой концентрации в кишечнике.
В отличие от глюкозы и галактозы фруктоза транспортируется системой, не зависящей от ионов Na+. На этом основана возможность лечения синдрома нарушенного всасывания глюкозы и галактозы.
Рис. 14.1. Схема переваривания углеводов [по Е.С. Северину, 2000].
Крахмал частично переваривается в полости рта под действием амилазы слюны, расщепляющей 1-4-гликозидные связи. Амилаза слюны инактивируется в желудке, так как оптимальное значение рН для ее активности составляет 6,7. Панкреатическая амилаза, расщепляющая 1-4-гликозидные связи, продолжает гидролиз крахмала в тонкой кишке, способствуя образованию дисахаридов мальтозы и изомальтозы. Далее мальтоза и изомальтоза вместе с другими пищевыми дисахаридами гидролизуются специфическими глюкозидазами на поверхности клеток кишечника (возможно, и внутри клеток) до соответствующих мономеров.
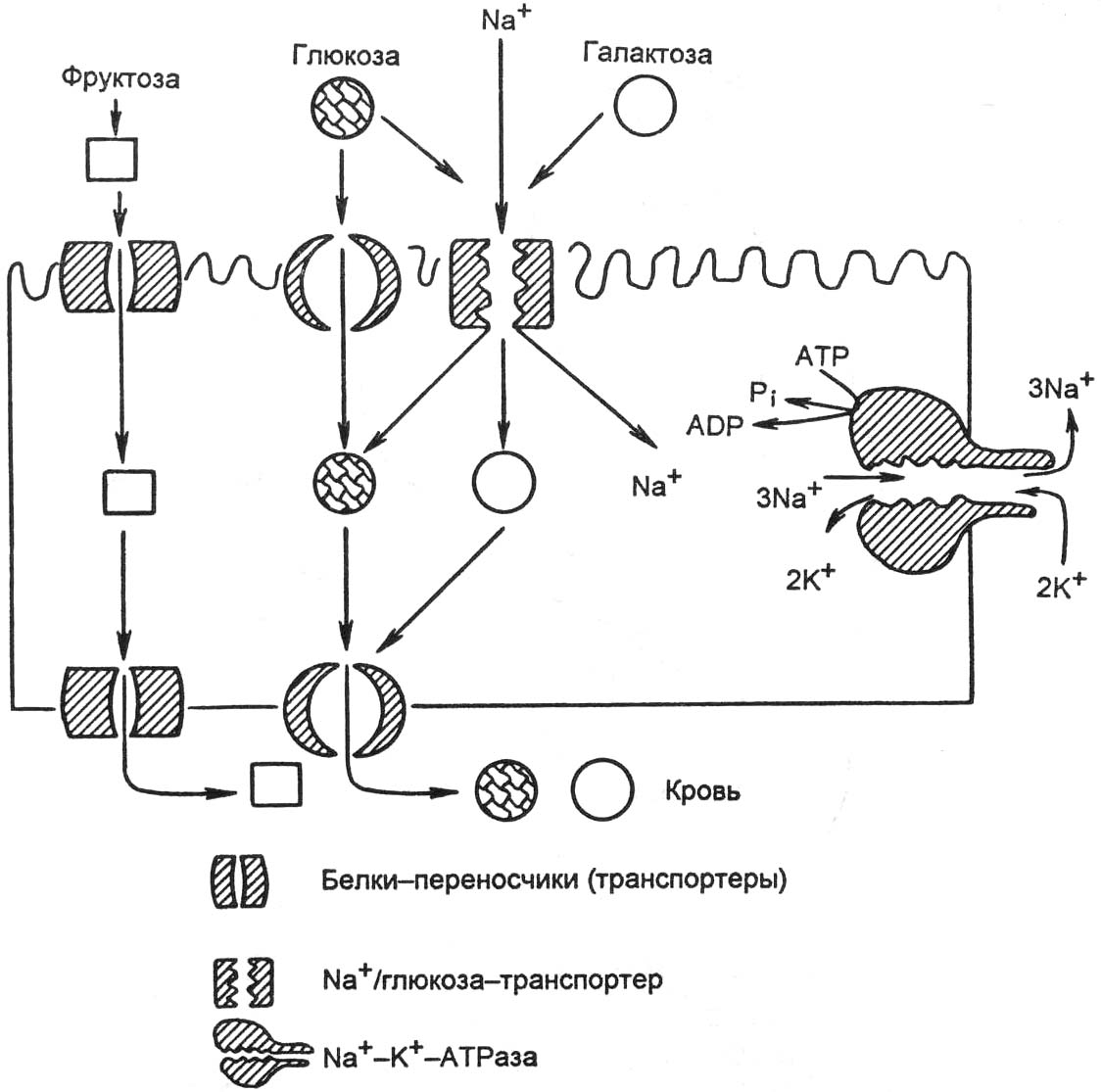
Рис. 14.2. Схема всасывания углеводов в кишечнике [по Dawn В., Marks, 1996], объяснение в тексте.
Из этих простых сахаров глюкоза по своей распространенности в качестве углеводного компонента пищи намного превосходит все остальные.
Поскольку внутри клеток свободная глюкоза как таковая практически отсутствует, вся поглощаемая тканями глюкоза подвергается следующим основным метаболическим превращениям:
накопление в виде гликогена;
окисление через анаэробный гликолиз (путь Эмбдена-Мейергофа) до пирувата и лактата;
окисление через цикл трикарбоновых кислот (цикл Кребса) или в меньшей степени через пентозный цикл до СО2;
превращение в жирные кислоты и накопление в виде триглицеридов;
высвобождение из клетки в виде свободной глюкозы.
Нарушения всасывания углеводов могут возникать при врожденной недостаточности специфического фермента или транспортной системы, необходимой для обмена определенного сахара. В том и другом случаях сахар накапливается в просвете кишечника, повышая осмолярность кишечного сока и тем дополнительно увеличивая всасываемость воды в просвет кишечника.
Общие признаки синдромов нарушения всасывания углеводов:
диарея,
вздутие живота после приема в пищу определенного сахара,
кислая реакция кала (pH<6,0) - поскольку отдельные углеводы метаболизируются бактериями толстого кишечника до органических кислот,
раздражительность,
отставание в росте,
отсутствие подъема сахара в крови после сахарной нагрузки,
дефект определенного фермента в слизистой оболочке кишечника.
Наследственная недостаточность сахаразы и изомальтазы проявляется, если в рацион ребенка добавляют сахарозу и крахмал, больные дети обычно неохотно едят сладкое.
Первичное нарушение всасывания глюкозы и галактозы встречается редко, проявляется вскоре после первого кормления профузной диареей, дегидратацией, ацидозом и гипогликемией.
Врожденная недостаточность лактазы. Гидролиз лактозы до глюкозы и галактозы опосредован лактазой кишечного эпителия. У детей с дефицитом этого фермента появляются стойкая диарея и гипотрофия. Нагрузка лактозой усиливает симптоматику. Безлактозная диета устраняет ее.
В эту же группу входят:
непереносимость сахарозы-изомальтозы,
непереносимость лактозы без недостаточности лактазы (биохимия синдрома неизвестна),
эссенциальная фруктозурия,
наследственная непереносимость фруктозы (дефицит фруктозо-1-фосфатальдолазы). В этом случае после поступления фруктозы через 30 мин начинается рвота, пот, диарея и даже кома;
недостаточность фруктозо-1,6-дифосфатазы.
В некоторых случаях встречается низкая активность лактазы вторичного характера, особенно при нарушении нейро-гуморальной регуляции, воспалении слизистой оболочки желудочно-кишечного тракта, отравлениях ядами типа монойодацетата и флоридзина, как следствие операций на ЖКТ.
Наследственные нарушения метаболизма фруктозы обусловлены дефектами ферментов: фруктокиназы, фруктозо-1-фосфатальдолазы и фруктозо-1,6-дифосфотазы.
Галактоземия - это результат нарушения обмена галактозы, обусловленное наследственным дефектом любого из трех ферментов, включающих галактозу в метаболизм (алактокиназа, галактозо-1-фосфатуридилтрансфераза (ГАЛТ), уридилфосфат-4-эпимераза).
Галактоземия вследствие недостаточности галактозил-1-фосфатуридилтрансферазы (ГАЛТ) известна наиболее хорошо. Заболевание проявляется очень рано, особенно тяжело протекает у детей, так как основным источником углеводов служит грудное молоко, содержащее лактозу. Ранними симптомами являются рвота, диарея, дегидратация, снижение массы тела, желтуха. Они появляются вскоре после рождения, как только ребенок начинает получать молоко.
В крови, моче и тканях повышается концентрация галактозы и галактозо-1-фосфата. В тканях глаза (в хрусталике) галактоза восстанавливается алдольредуктазой с образованием галактитола (дульцит). Восстановление галактозы характерно и для нормального метаболизма, но протекает с небольшой скоростью. При галактоземии галактитол накапливается в стекловидном теле и связывает большое количество воды. Нарушается баланс электролитов, а чрезмерная гидратация хрусталика приводит к развитию катаракты, которая наблюдается уже через несколько дней после рождения. Катаракта может быть обнаружена только с помощью специальных методов и не определяется при помощи простого офтальмоскопа. Серьезные изменения наблюдаются в печени в связи с накоплением галактозо-1-фосфата. Нарушаются функции печени и почек. Выявляются нарушения в клетках полушарий большого мозга и мозжечка, в тяжелых случаях - отек мозга, задержка умственного развития; возможен летальный исход.
Первой внутриклеточной реакцией, в которой участвует глюкоза, является ее фосфорилирование в глюкозо-6-фосфат гексокиназой и глюкокиназой. Нарушение этого процесса также неблагоприятно сказывается на всасывании углеводов.
Дальнейший метаболизм глюкозо-6-фосфата идет по одному из 4-х возможных путей:
Аэробный гликолиз (основной).
Анаэробный гликолиз.
Пентозофосфатный цикл.
Синтез гликогена.
Аэробный гликолиз проходит 3 этапа:
Образование пирувата.
Окислительное декарбоксилирование пирувата (включает образование АцКоА).
Цикл трикарбоновых кислот (цикл Кребса), в который включается АцКоА и щавелево-уксусной кислоты (ЩУК), образуя лимонную кислоту. В конце из каждой молекулы глюкозы образуется 6 молекул СО2.
На этапах метаболизма глюкозы запускается цепь ферментов тканевого дыхания, первым из которых является НАД-зависимая дегидрогеназа. Дыхательная цепь срабатывает 10 раз за цикл, перенося 2Н+ на внешнюю сторону митохондриальной мембраны, а 2 электрона - на внутреннюю, что сопровождается выделением энергии, достаточной для синтеза 3-х молекул АТФ, т.е. всего 30 молекул АТФ за цикл.
Переход ФАД в ФАД Н2 дает укороченную цепь тканевого дыхания с 4-мя молекулами АТФ. Этот процесс, окислительное фосфорилирование сопряженное с дыханием, дает всего 34-е молекулы АТФ. Субстратное фосфорилирование еще 6 молекул АТФ.
Итого 40 молекул АТФ минус 2 молекулы (затраченные на фосфорилирование глюкозы) = 38 молекул АТФ и 6 молекул СО2 на 1-ую молекулу глюкозы. КПД около 44%.
Рис. 14.3. Цикл трикарбоновых кислот (цикл Кребса) [по Т.Т. Березову, Б.Ф. Коровкину, 1998].
Рис. 14.4. Образование 38 молекул АТФ в результате окисления 1 молекулы глюкозы [по Б. Гринстейн, А. Гринстейн, 2000].
Анаэробный гликолиз совпадает до стадии пирувата. Далее вместо окислительного декарбоксилирования пируват подвергается восстановлению, приняв на себя 2Н+ от дегидрогенозы НАД Н2 с образованием лактата (молочной кислоты). Катализирует лактатдегидрогеназа. В эритроцитах только анаэробный гликолиз и большое значение в работающих мышцах. Эффективность - 2-е молекулы АТФ на 1-у молекулу глюкозы.
Рис. 14.5. Последовательность реакций гликолиза [по Т.Т. Березову, Б.Ф. Коровкину, 1998]. 1 - гексокиназа; 2 - фосфоглюкоизомераза; 3 - фосфофруктокиназа; 4 - альдолаза; 5 -триозофосфатизомераза; 6 - глицеральдегидфосфатдегидрогеназа; 7 -фосфоглицераткиназа; 8 - фосфоглицеромутаза; 9 - енолаза; 10 - пируваткиназа; 11 - лактатдегидрогеназа.
Пентозофосфатный путь - окислительная ветвь образует 2 молекулы НАДФ Н2 (необходимый для биосинтеза жирных кислот, холестерина и т.д.). В неокислительной ветви – рибозо-5-фосфат, который используется для синтеза РНК, ДНК, АТФ, КоА, НАД и ФАД.
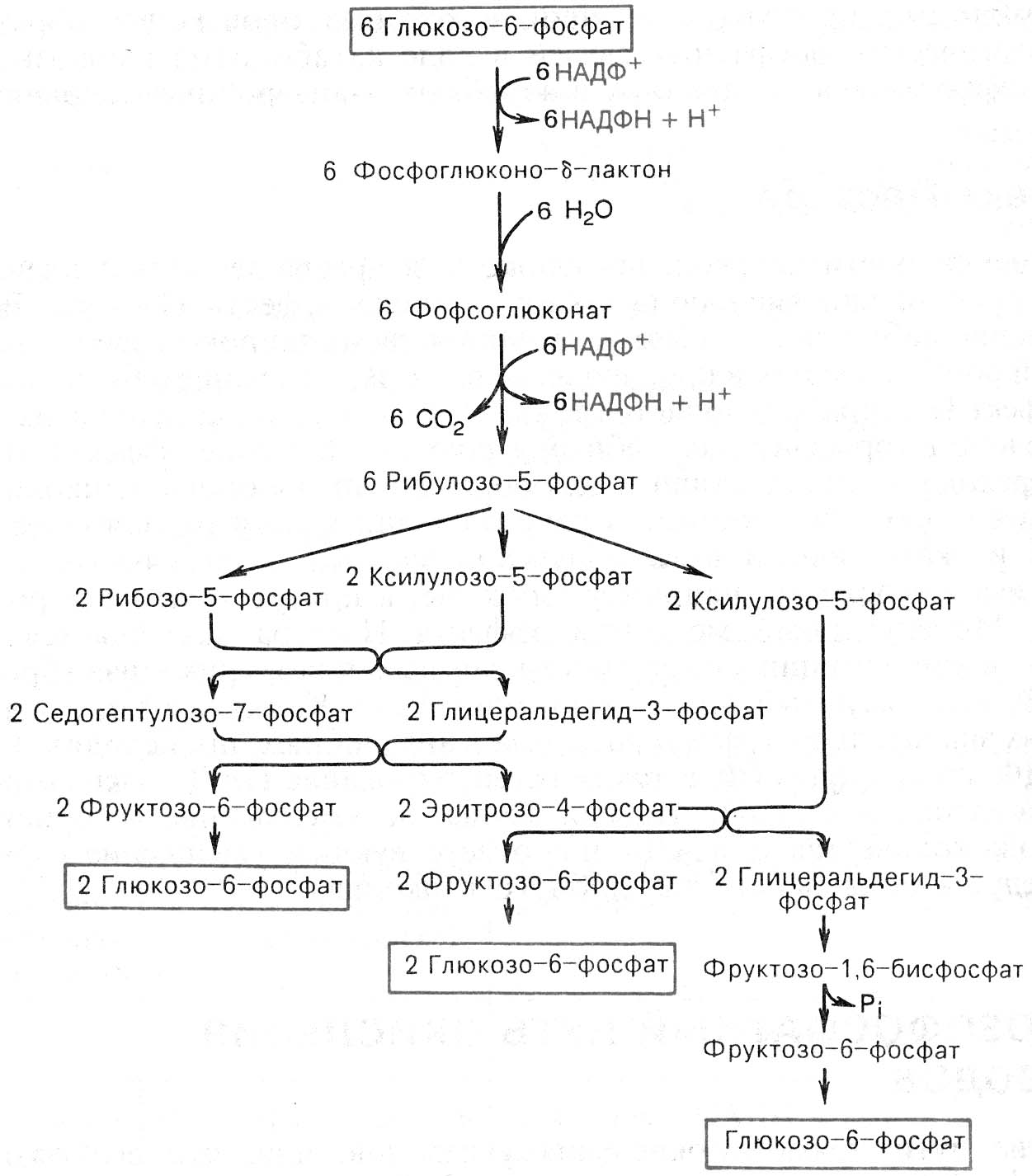
Рис. 14.6. Схема пентозофосфатного пути метаболизма углеводов [по Т.Т. Березову, Б.Ф. Коровкину, 1998].
Глюконеогенез. Синтез глюкозы из неуглеводных продуктов; в первую очередь лактат и пируват, гликогенные аминокислоты, глицерол и ряд др. соединений. Т.е. предшественниками глюкозы может быть пируват или любое соединение, превращающееся в процессе катаболизма в пируват или один из промежуточных продуктов цикла Кребса.
Нарушения депонирования углеводов. После приема пищи большая часть глюкозы, метаболизирующейся в печени, превращается в гликоген, который при первой необходимости служит готовым источником глюкозы. Однако общее содержание его в печени довольно ограничено (в среднем 70-100 г) и способно обеспечить потребности организма в глюкозе в течение не более 8-12 часов.
Реакция образования гликогена зависит от активности гликогенсинтетазы, которая, в свою очередь, находится в обратной в зависимости от внутриклеточного уровня цАМФ.
До сих пор нет ясности в вопросе, опосредована ли активность гликогенсинтетазы главным образом гормонами (например, инсулином, глюкагоном или адреналином - первый ее повышает, два остальных - понижают) или субстратом, т.е. глюкозой.
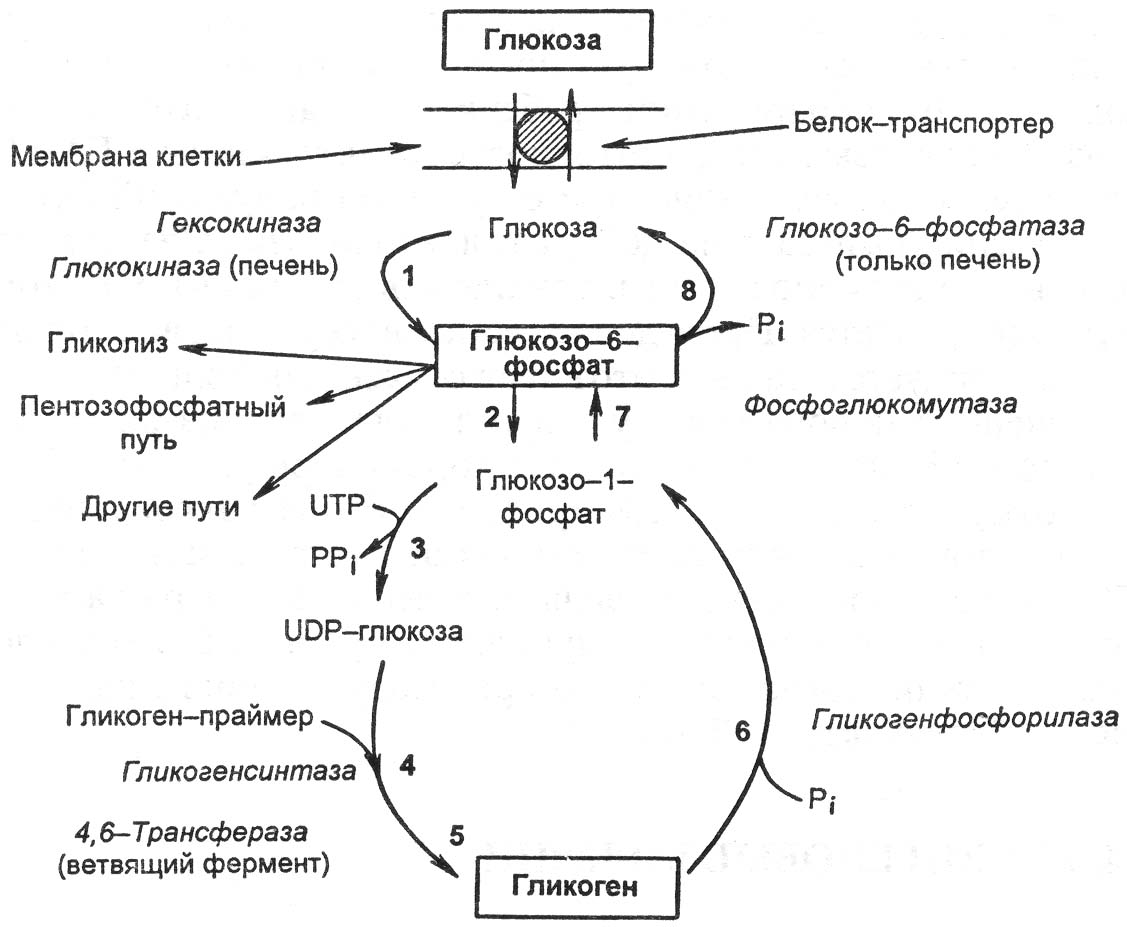
Рис. 14.7. Синтез и распад гликогена [по Е.С. Северину, 2000]. 1, 2, 3, 4, 5 - реакции синтеза гликогена; 6, 7, 8 - реакции распада гликогена.
Снижение синтеза гликогена отмечается при миастении, гипоксии, тогда как повышенный распад наблюдается при охлаждении, перегревании, боли, судорогах, эмоциональном стрессе.
Выделяют т.н. агликогеноз - наследственное заболевание, вызванное дефектом гликогенсинтетазы. В печени почти или полностью отсутствует гликоген, выражена гипогликемия.
Следует сказать, что гликогенолиз контролируется ферментом фосфорилазой, которая, подобно гликогенсинтетазе, существует в неактивной форме и должна активироваться.
Гликогенолиз в печени и мышечной ткани приводит к образованию различных продуктов: в печени - к образованию свободной глюкозы, в мышцах - к высвобождению лактата и пирувата, поскольку глюкозо-6-фосфат не может превращаться в глюкозу, а вступает на гликолитический путь.
Гликогенолиз может рассматриваться как средство адаптации только к острым потребностям организма в глюкозе. В условиях длительного дефицита глюкозы (голодание, нарушение реабсорбции глюкозы в почках, диабет) в ход идет другой, более продолжительный механизм – глюконеогенез. Единственной другой тканью (помимо печени), в которой возможен глюконеогенез и превращение глюкозо-6-фосфата в глюкозу, является корковый слой почек.
Особую группу представляют болезни накопления гликогена, или гликогенозы. В основе этой патологии лежит энзиматический дефект, который проявляется необычной структурой гликогена или его избыточным накоплением в печени, почках, нервной системе. Характерны гепатоспленомегалия, задержка роста. Наиболее часто встречаются 6 типов гликогенозов.
I тип (гликогеноз Гирке) - следствие дефицита глюкозо-6-фосфатазы. Встречается наиболее часто, проявляется гипогликемией, накоплением гликогена в печени и почках, ацидозом (за счет накопления лактата) и гепатоспленомегалией. Больные отличаются малым ростом.
II тип (гликогеноз Помпе) - обусловлен дефектом кислой альфа-1,4-глюкозидазы. Отличается от других гликогенозов тем, что дефектным становится лизосомальный фермент. Проявляется генерализованным накоплением гликогена, поражением печени, почек, нервной системы, гипертрофией миокарда. Болезнь быстро прогрессирует и никакое лечение не в состоянии предотвратить смерть больного.
III тип (лимитдекстриноз, болезнь Кори, Болезнь Форбса) - вызывается дефицитом амило-1,6-гликозидазы. Больным свойственны гепатомегалия, мышечная, слабость, гипогликемия натощак, "кукольное личико". Течение относительно доброкачественное.
IV тип (амилопектиноз, болезнь Андерсена) - редко встречающаяся тяжелая форма гликогенозов. Для нее характерен цирроз печени с желтухой и печеночной недостаточностью, развивающийся в грудном возрасте. Отложение гликогена генерализованное, гликоген структурно изменен с очень длинными наружными ветвями. До сих пор не предложено никакого лечения, кроме симптоматического.
V тип (недостаточность миофосфорилазы, болезнь Мак-Ардла) - вызван дефицитом фосфорилазы, активирующей бета-киназу в мышцах и печени. Интересна история этого заболевания. Первый случай был расценен как психосоматическое нарушение. У больного в покое отсутствовали какие бы то ни было симптомы, но даже после умеренной нагрузки возникали боли в мышцах. Первые проявления болезни возникают обычно в 25-30 лет. Печень не поражается, структура гликогена нормальна, нет смертельных исходов, т.к. гамма-амилаза совместно с амило-1,6-гликозидазой расщепляют гликоген до глюкозы. Единственный признак - миастения, особенно после физической активности.
VI тип (недостаточность печеночного фосфорилазного комплекса, болезнь Херса) - дефект печеночной фосфорилазы, ведущий и избыточному накоплению нормального гликогена в печени. Отмечают гепатомегалию, легкое замедление темпов роста. Прогноз для жизни хороший, умственное развитие нормальное.
Достаточно редко встречается гликогеноз VII типа (дефект мышечной фосфофруктокиназы, болезнь Терье), схожий с болезнью Мак-Ардла и проявляющийся нарастанием уровня лактата и пирувата в крови после мышечной работы.
Этиология и патогенез гипогликемических состояний.
Регуляция уроня глюкозы в крови
В норме уровень глюкозы в плазме крови колеблется в пределах 3,3-5,5 мМоль/л – нормогликемия. Снижение ниже 3,3 мМоль/л – гипогликемия. Повышение выше 5,5 мМоль/л - гипергликемия. Снижение менее 2,7 мМоль/л - гипогликемическая кома. Увеличение более 8,8 мМоль/л – глюкозурия. Увеличение более 22 мМоль/л - гипергликемическая кома.
Инсулин полипептидный гормон, состоящий из 2-х полипептидных цепей, обозначаемых как А- и В-цепи, соединенные двумя дисульфидными мостиками. Полная молекула содержит 51 аминокислоту с молекулярной массой 5800.
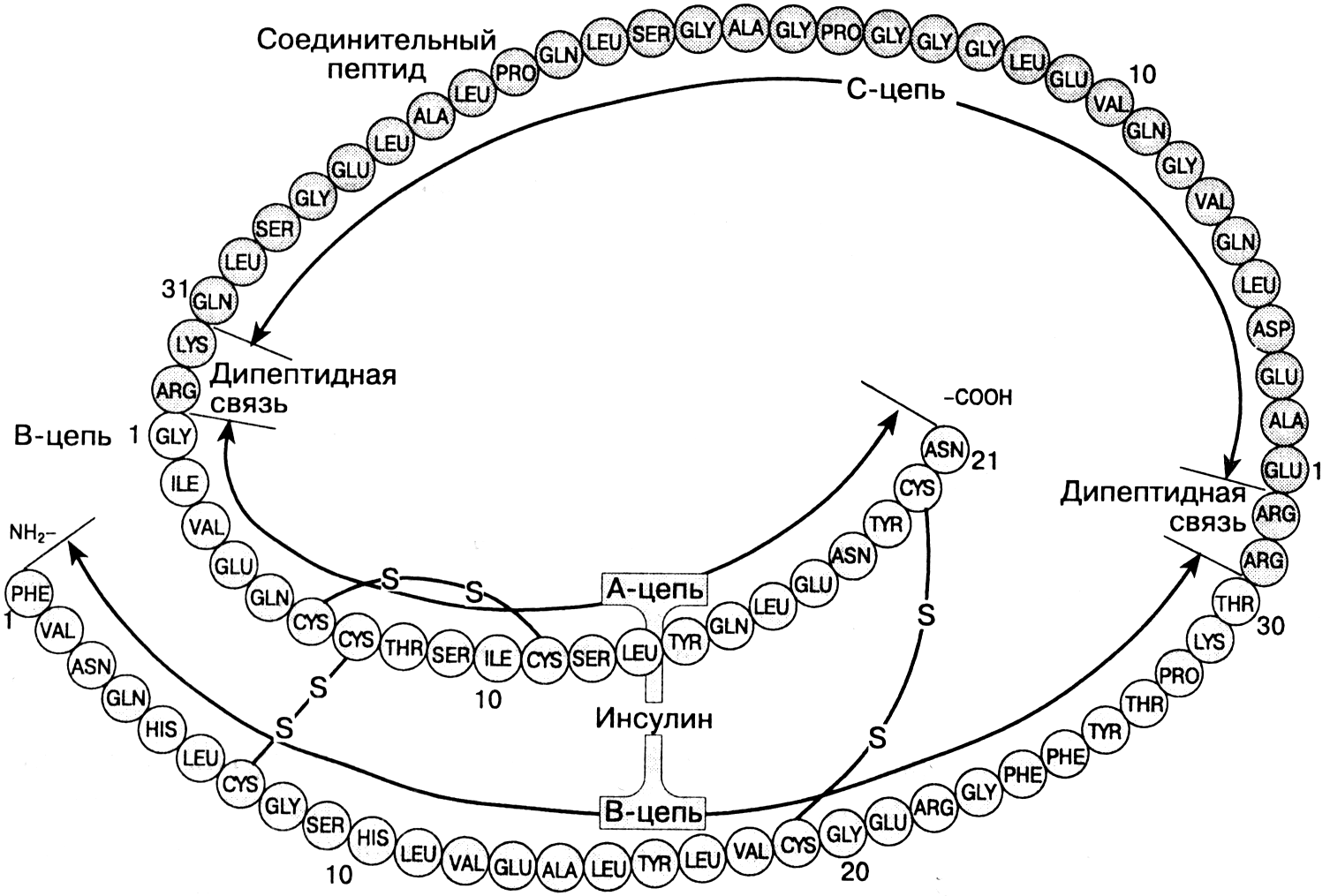
Рис. 14.8. Молекулы проинсулина и инсулина. Показаны последовательность аминокислот в молекуле проинсулииа, А- и В-цепи молекулы активного инсулина. Расщепление дипептидных связей ведет к образованию молекулы инсулина и неактивного С-пептида [по В.М. Кэттайл, Р.А. Арки, 2001].
Инсулин синтезируется бета-клетками в виде одноцепочечного предшественника – проинсулина (путем отщепления С-пептида), а тот, в свою очередь, из препроинсулина.
К стимуляторам синтеза и секреции инсулина относятся: глюкоза, манноза, лейцин, СТГ, глюкагон, глутаминовая кислота, секретин. Адреналин ингибирует синтез инсулина.
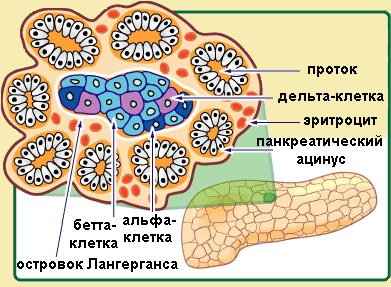
Рис. 14.9. Строение островкового аппарата поджелудочной железы.
В нормальных островках бетта-клетки составляют 60%, альфа-клетки - 25% (глюкагон) и дельта-клетки - 10% (соматостатин). Островок Лангерганса представляет собой маленький орган, все клетки которого координированно отвечают на стимулы извне и собственные паракринные влияния.
Главная функция инсулина регуляция метаболизма белков, жиров, углеводов. Это анаболический гормон. Суммарно инсулин снижает уровень глюкозы в крови.
Его эффекты на мышцы, печень и жировую ткань:
стимуляция захвата клетками глюкозы, аминокислот, жирных кислот;
усиление синтеза гликогена (гликогеногенез в печени и мышцах);
усиление синтеза жиров - липогенез (ожирение при 2 типе СД), есть сведения, что тормозит липазу, но при 2 типе СД ожирение по андроидному типу (жир легко запасается и легко расщепляется), т.к. активируется липогенез и не тормозится липолиз;
усиление синтеза белков (протеиногенез);
стимуляция гликолиза;
торможение глюконеогенеза и распада гликогена (гликогенолиза), белков и триглицеридов.
50% инсулина распадается в печени под действием инсулиназы, поэтому при патологии печени развивается гиперинсулинемия.
Контринсулярные гормоны (увеличивают уровень глюкозы в крови);
Глюкагон (-клетки островков Лангерганса поджелудочной железы):
ускоряет гликогенолиз и глюконеогенез в печени;
тормозит синтез белка и ускоряет протеолиз.
Глюкокортикостероиды (глюкокортикоиды) и актг:
ускоряют глюконеогенез;
активируют гликогенолиз.
Адреналин:
активирует гликогенолиз,
активирует липолиз (ЖК - антагонисты инсулина)
СТГ:
активирует инсулиназу,
конкурирует с инсулином за рецепторы.
Соматостатин:
угнетает синтез инсулина и гликогена.
Гипогликемия – снижение концентрации глюкозы в плазме ниже нижней границы нормы.
Гипогликемия представляет собой снижение уровня глюкозы плазмы до таких значений, когда появляются клинические симптомы, исчезающие после нормализации снижения глюкозы.
Скорость развития гипогликемии зависит от пола больного, быстроты снижения уровня глюкозы, исходного содержания глюкозы.
В среднем принято считать, что гипогликемия развивается при снижении уровня глюкозы до 2,5-3,0 ммоль/л.
Классификация, основанная на характеристике условий, в которых развивает гипогликемия. С этих позиций можно выделить 3 основные виды гипогликемий:
I - гипогликемия натощак:
а) инсулинпродуцирующая опухоль бетта-клеток;
б) гипогликемия поврежденных с кетозом.
II - гипогликемия после еды:
а) спонтанная реактивная гипогликемия;
б) ранние стадии сахарного диабета.
III - индуцированная гипогликемия:
а) алкогольная гипогликемия;
б) передозировка инсулина.
Гипогликемия натощак характеризуется неэффективностью поддержания нормального уровня глюкозы в условиях воздержания от пищи.
В состоянии натощак глюкоза в организме человека потребляется почти исключительно мозгом. Небольшие количества ее потребляются также форменными элементами крови. Совершенно иная ситуация складывается в инсулинзависимых тканях, таких, как мышцы и жировая ткань, которые практических не потребляют глюкозу при голодании, длящемся 12 ч и более. Если голодание продолжается более 12-14 часов, то включаются дополнительные механизмы поддержания нормогликемии. Мозг утилизирует глюкозу со скоровстью 125 г/сут, поэтому запасов гликогена в печени явно недостаточно. На помощь приходит глюконеогенез и липолиз с последующим окислением жирных кислот и прогрессивным повышением в плазме уровня кетоновых тел (бета-оксимасляная кислота, ацетон и ацетоуксусная кислота). При более длительном голодании (дни и недели) в ход идут запасы белка, что неминуемо приводит к смерти (если вовремя не остановиться).
Таким образом, поддержание нормогликемии в состоянии натощак зависит от 3-х основных факторов:
гормональной среды, характеризующейся исходным или сниженным уровнем инсулина и исходным или повышенным уровнем глюкагона, СТГ и кортизола;
печени, в которой не нарушены процессы гликогенолиза и глюконеогенеза;
субстратов процессов глюконеогенеза.
Следовательно, гипогликемию натощак можно подразделить на эндокринную, печеночную и субстратную.
Итак, гипогликемия натощак подразделяется:
1. Эндокринная
а) Избыток инсулина или инсулиноподобных факторов:
островковоклеточные опухоли, продуцирующие инсулин;
внепанкреатические опухоли, вызывающие гипогликемию.
б) Дефицит СТГ:
гипопитуитаризм;
изолированный дефицит СТГ.
в) Дефицит кортизола:
гипопитуитаризм;
изолированный дефицит АКТГ;
Аддисонова болезнь.
2. Печеночная
а) болезни накопления гликогена;
б) дефицит ферментов глюконеогенеза;
в) острый некроз печени:
отравления;
вирусный гепатит.
г) застойная сердечная недостаточность.
3. Субстратная
а) гипогликемия беременных;
б) гипогликемия новорожденных с кетозом;
в) уремия;
г) алиментарная недостаточность.
4. Прочие причины
а) аутоиммунная инсулиновая гипогликемия.
Островковоклеточные опухоли поджелудочной железы вызывают гипогликемию за счет гиперпродукции инсулина. Состоит опухоль из гроздьев типичных бета-клеток. Патогенез гипогликемии у таких больных сводится либо к абсолютному повышению уровня инсулина в плазме натощак или во время физической работы, либо к отсутствию снижения уровня инсулина, которое в таких условиях происходит в норме.
Гипогликемия при дефиците гормонов обусловлена снижением или отсутствием их контринсулярного эффекта.
Гипогликемия при заболеваниях печени была частично нами уже рассмотрена (гликогенозы). Следует отметить, что это состояние встречается, и нередко, при вирусных гепатитах и тяжелых токсических поражениях печени.
Помимо снижения уровня инсулина, присутствия регуляторных гормонов и сохранности глюконеогенеза в печени, продукция глюкоза натощак требует и присутствия субстратов-предшественников, особенно аланина. У здоровых лиц скорость высвобождения аланина из мышц определяет скорость глюконеогенеза при голодании.
Гипогликемия после еды, или реактивная гипогликемия - это состояние можно определить как уменьшение содержания глюкозы в плазме в период перехода от состояния сытости к состоянию голода, достаточное для появления субъективных жалоб. Предполагают, что гипогликемия после еды может быть обусловлена либо отсутствием адекватного снижения утилизации глюкозы по мере уменьшения уровня глюкозы в плазме, либо неадекватностью поглощения глюкозы печенью и периферическими тканями.
В этой группе выделяют
идиопатическую (функциональную) гипогликемию,
алиментарную гипогликемию,
гипогликемию на ранних стадиях сахарного диабета.
Идиопатическая (функциональная) гипогликемия встречается чаще у женщин 25-35 лет, больные внешне не отличаются от здоровых. Жалобы достаточно неспецифичны - тошнота, слабость, сердцебиение. Симптомы эти могут существовать годами, не прогрессируя.
Алиментарная гипогликемия наблюдается иногда у больных с операциями на ЖКТ. Гипогликемия выражена больше, чем у больных с идиопатической гипогликемией. Считают, что гипогликемия у них обусловлена кишечными, а не панкреатическими дефектами.
Давно известно, что гипогликемия после еды может быть проявлением инсулиннезависимого СД (II типа, диабета взрослых). У таких больных натощак уровень глюкозы находится в пределах нормы, но рано повышается вследствие недостаточной секреции инсулина и отмечается поздняя гиперинсулинемия.
Из других причин гипогликемию после еды наблюдали при ожирении, почечной глюкозурии и различных гормондефицитных состояниях.
Индуцированные гипогликемии возникают при:
передозировке инсулина и других ССП;
приеме алкоголя;
врожденных дефектах метаболизма.
Алкогольная гипогликемия распространена среди пьющих, но мало или совсем не закусывающих лиц. Синдром развивается спустя 6-24 часа после алкогольного эксцесса и поэтому запах алкоголя может не ощущаться.
Механизм развития алкогольной гипогликемии, вероятно, заключается в уменьшении продуцирования глюкозы печени из-за угнетения алкоголем глюконеогенеза (снижение использования лактата и аланина).
Клинические проявления гипогликемии обусловлены двумя факторами: 1) снижением уровня глюкозы в головном мозге (нейрогликопения); 2) стимуляцией симпатоадреналовой системы.
Нейрогликопения проявляется головной болью, утомляемостью, помрачением сознания, галлюцинациями, судорогами и комой.
Симптомы адренергической стимуляции включают сердцебиение, возбуждение, потливость, дрожь и чувство голода. Они появляются раньше, чем все прочие, предупреждая о надвигающейся коме. Приступ можно оборвать, приняв глюкозу или углеводсодержащую пищу.
Лечение гипогликемии заключается в замедленном введении 50-70 мл 40% раствора глюкозы, можно повторно.
39. – Сахарные диабеты. Виды. Этиология, сходства и отличия патогенеза различных видов сахарных диабетов. Диагностические критерии.
Классификация диабетов
Сахарный диабет (общее важнейшее звено – гипергликемия)
Юношеский, I тип, инсулинзависимый (основное звено патогенеза – недостаток инсулина).
Старческий (пожилых), II тип, инсулинонезависимый, (инсулинорезистентный) (основное звено патогенеза – дефект рецепторов к инсулину в клетках).
Несахарный диабет
Центральный или гипоталамический (недостаток АДГ).
Почечный (дефект рецепторов клеток почечных канальцев к АДГ)
Почечный (болезнь Фанкони, почечная глюкозурия) - ферментопатия в клетках почечных канальцев с нарушением реабсорбции глюкозы.
Сахарный диабет представляет собой хроническое нарушение всех видов обмена веществ (преимущественно углеводного), обусловленное абсолютной или относительной инсулиновой недостаточностью и характеризующееся стойкой гипергликемией.
Сахарным диабетом страдают около 2% всего населения Земли и с каждым годом эта цифра увеличивается.
Этиология сахарного диабета. Считают, что он представляет собой гетерогенную группу расстройств с различной этиологией.
Основными идентифицированными этиологическими факторами являются:
Наследственность.
Можно утверждать, что генетические факторы играют роль в развитии всех клинических форм спонтанного диабета, но каждый из них характеризуется специфическим способом наследования, чаще всего это полигенное наследование.
Предрасположенность к диабету I типа связана с локусом HLA-D на коротком плече 6-й хромосомы. Этот участок обусловливает иммунологические реакции. Повреждение генов этой области создает предрасположенность к аутоиммунной деструкции бета-клеток, вызываемой факторами внешней среды, сочетание которых в каждом отдельном случае может быть различным.
Аутоиммунные процессы.
У больных инсулинзависимым сахарным диабетом часто обнаруживаются антитела к белкам бета-клеток, что указывает на аутоиммунный компонент патогенеза заболевания. Эти антитела принадлежат к классу Ig G и являются органоспецифическими.
Вирусные инфекции.
Бета-клетки могут избирательно поражаться бета-тропными вирусами (типа Коксаки, кори, эпидемического паротита и т.д.), что подтверждается сезонными колебаниями частоты развития сахарного диабета I типа, прямой передачей сахарного диабета от человека экспериментальным животным и гистоморфологической картиной на посмертной аутопсии.
Питание.
Обращает внимание частое сочетание сахарного диабета и ожирения. Особенно эта связь характерна для диабета II типа. У генетически предрасположенных лиц с ограниченной способностью секретировать инсулин развитие ожирения создает такие потребности в гормоне, которые превышают секреторную способность бета-клеток и приводят к развитию сахарного диабета.
Таблица 14.1.
Дифференциальная диагностика диабетов
Признак |
Сахарный |
Несахарный |
Почечный (Фанкони) |
||
I тип |
II тип |
Гипоталамический |
Почечный |
||
Полиурия |
+ |
+ |
+ |
+ |
+ |
Полидипсия |
+ |
+ |
+ |
+ |
+ |
Гипергликемия |
+ |
+ |
- |
- |
- |
Глюкозурия |
+ |
+ |
- |
- |
+ |
Инсулин |
< |
N |
N |
N |
N |
С-пептид |
< |
N |
N |
N |
N |
Тест толерантности к глюкозе |
< |
< |
N |
N |
N |
АДГ |
- |
- |
< |
- |
- |
40. - Патогенез сахарного диабета 1 и 2 типа. Изменения белкового, липидного и водно-солевого обмена при сахаоном диабете. Диабетические комы. Понятие метаболического Х-синдрома.
Патогенез сахарного диабета. Основой, вокруг развертываются все звенья сахарного диабета, является дефицит инсулина.
Гипергликемия вызвана нарушением транспорта глюкозы в клетки.
Чувство жажды (полидипсия) обусловлена гиперосмолярностью крови, дегидратация клеток.
Глюкозурия и полиурия развиваются после преодоления почечного порога для глюкозы (8,8 мМоль/л).
Полифагия вызвана энергодефицитом.
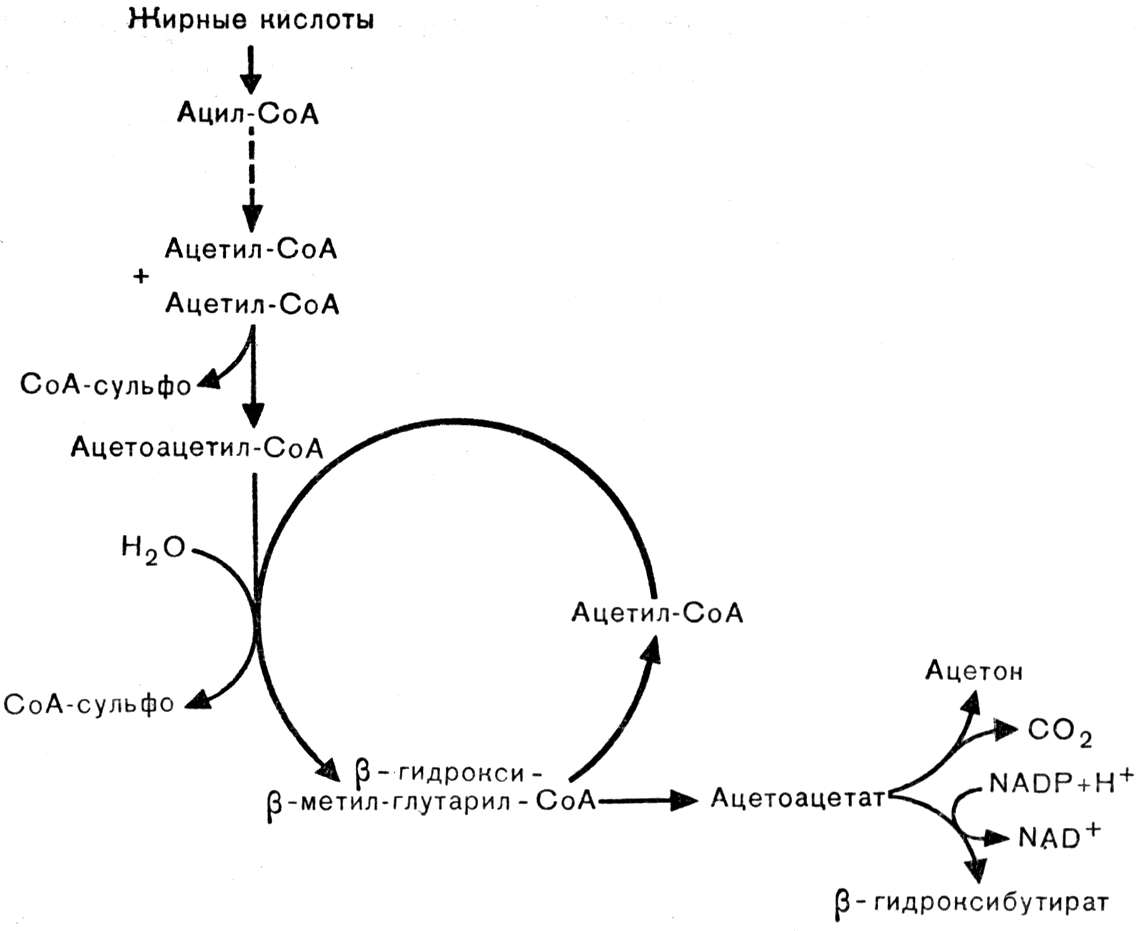
Рис. 14.10. Механизм образования кетоновых тел. Непрерывными линиями обозначены реакции образования кетоновых тел, пунктиром - этапы превращения ацил-КоА в ацетил-КоА [по А.Г. Мазовецкому, В.К. Великову, 1987].
Кетонемия и кетонурия обусловлена «перекосом» в сторону использования в качестве источника энергии липидов. Образующийся при этом Ацетил-КоА не сгорает полностью в ЦТК («жиры сгорают в пламени углеводов»), и часть его идет на синтез кетоновых тел → из кетоновых тел синтезируется холестерин (атеросклероз).
Нарушение кислотно-основного состояния в виде ацидоза связаны с кетоацидозом
Отрицательный азотистый баланс возникает вследствие глюконеогенеза из аминокислот.
Гиперосмотическая дегидратация - в связи с выделением с мочой глюкозы, кетоновых тел, азотсодержащих соединений, Na+. Клеточная дегидратация (мозг диабетическая кома) из-за гиперосмолярности плазмы.
Накопление гликозилированного гемоглобина (гликогемоглобина). Это гемоглобин, в котором молекула глюкозы конденсируется с бета-концевым валином бета-цепи молекулы гемоглобина А гемическая гипоксия.
Диабетическая микроангиопатия чаще всего проявляются поражениями капилляров почек и сетчатки глаза. В основе этих состояний лежат биохимические изменения при сахарном диабете. Базальная мембрана клубочков при сахарном диабете содержит избыточное количество гликопротеинов. Более того, в почках возрастает активность фермента глюкозилтрансферазы, ответственной за модификацию гликопротеинов. Избыточное гликозилирование белков (гемоглобина, альбумина и других) приводит к их повышенному отложению в микроциркуляторном русле, что и проявляется в виде микроангиопатии.
Диабетическая нейропатия может затрагивать деятельность практически любой системы организма и имитирует многочисленные неврологические заболевания. В процесс могут вовлекаться чувствительные, двигательные и вегетативные нервы. Нейропатия может протекать с демиелизацией волокон или без нее. Вегетативная дисфункция при сахарном диабете, как правило, проявляется постуральной гипотензией, импотенцией, нарушениями функции ЖКТ и т.д. Несомненно, что свой вклад вносит сорбитоловый путь метаболизма глюкозы. Гипергликемия активирует его, что приводит к накоплению сорбитола и фруктозы и тем самым к осмотическим сдвигам во внутриклеточной среде. Последнее сопровождается гипоксией нервных клеток и нарушением их функции.
Нарушение нервной проводимости при сахарном диабете может быть связано со снижением уровня миоинозитола в нервной ткани. Это циклический гекситол, синтезируемый в нервной ткани из глюкозы и необходимый для синтеза фосфолипидов клеточной мембраны.
Показано, что при сахарном диабете нарушается аксоплазматический ток, т.е. транспорт белков и нейромедиаторов из тела клетки по аксонам различной длинны.
Нефропатия – нарушение кровоснабжения почек.
Нарушения обмена веществ, которые инициирует дефицит инсулина при сахарном диабете.
Углеводный обмен: снижается способность печени утилизировать глюкозу, резко повышается гликогенолиз и глюконеогенез (последний за счет активации цикла Кори, связывающего его с гликолизом), снижается активность цикла Кребса и пентознофосфатного окисления глюкозы, зато возрастает использование глюкозы в биосинтезе гликопротеинов и сорбитоловом пути окисления. Последние представляют альтернативные пути метаболизма глюкозы, имеющие важное значение в патогенезе осложнений сахарного диабета.
Жировой обмен: сахарный диабет сопровождается значительным "опустошением" жировых депо, т.е. активацией липолиза (процесс, который контролируется инсулинзависимой липазой). В результате может возникнуть вторичная гипертриглицеридемия. В печени увеличивается содержание жиров, большую часть которых она способна окислять только до уровня ацетил-КоА. Затем двухуглеродные фрагменты образуют ацетоуксусную, бета-оксимасляную кислоту и ацетон.
Эти вещества и носят собирательное название кетоновых тел. Присутствуя в избыточных количествах, они усугубляют метаболический ацидоз, ацетон же способствует появлению у больного характерного фруктового запаха при дыхании. На фоне повышения продукции кетоновых тел увеличивается образование триглицеридов в печени, что приводит к ее жировой дистрофии. Образуемые в печени ацетоацетат и бета-оксимасляная кислота попадают в кровь и циркулируют в отношении 1:3. Они подвергаются окислению в мышечной ткани и частично утилизируются клетками мозга как альтернативный глюкозе энергосубстрат.
Атеросклероз у больных сахарным диабетом развивается раньше обычного. Более того, принято считать, что сахарный диабет сопровождается ускоренным старением организма. Предполагают, что атеросклероз при этом может быть усилен по крайней мере тремя путями:
под действием избыточного количества СТГ может усиливаться пролиферация гладкомышечных клеток;
повышенный синтез тромбоксана способствуют адгезии тромбоцитов и выделению митогена;
при диабете как одно из проявлений характерной липемии повышен уровень ЛПНП и снижению содержания ЛПВП. В результате пагубный эффект ЛПНП усиливается.
Рядом с геном инсулина обнаружен участок ДНК, состоящий из 2500 пар оснований (U-аллель), который служит постоянным генетическим маркером предрасположенности к атеросклерозу не только при типе II, но при типе I, а также у лиц без диабета. Таким образом, предрасположенность к атеросклерозу может быть генетической и реализоваться у больных сахарным диабетом чаще, чем у лиц без него.
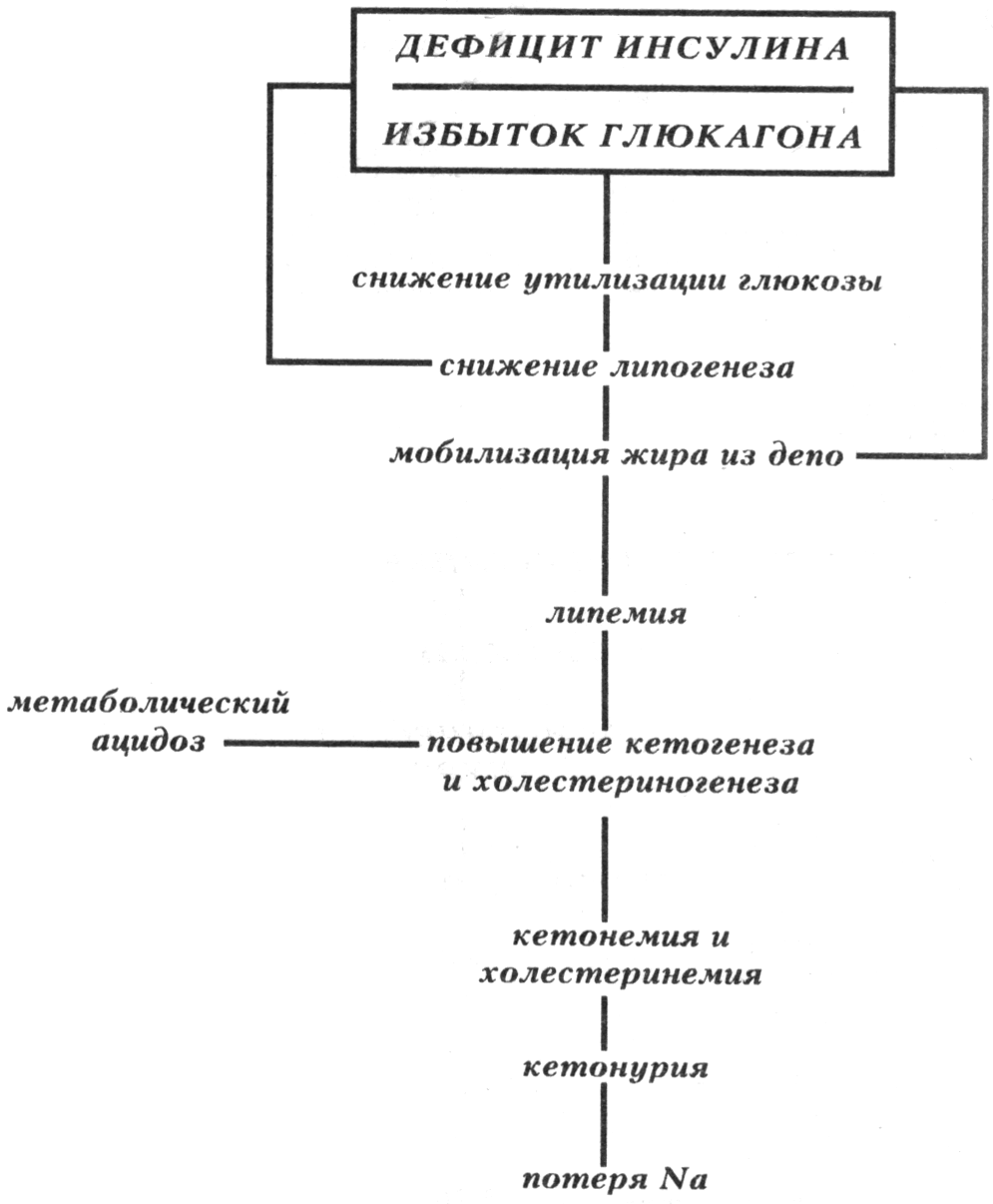
Рис. 14.11. Липидный обмен при сахарном диабете. Пояснения в тексте [по А.Ш. Зайчику, Л.П. Чурилову].
Белковый обмен: снижается синтез белка и повышается его катаболизм, прежде всего в инсулинчувствительных тканях (мышцах). Этот процесс сопровождается потерей организмом азота, а также выходом К+ и других внутриклеточных ионов в кровь с последующей экскрецией К+ с мочой.
Таким образом, дефицит инсулина характеризуется невозможностью восстановления или увеличения запасов энергетических веществ в организме при потреблении пищи.
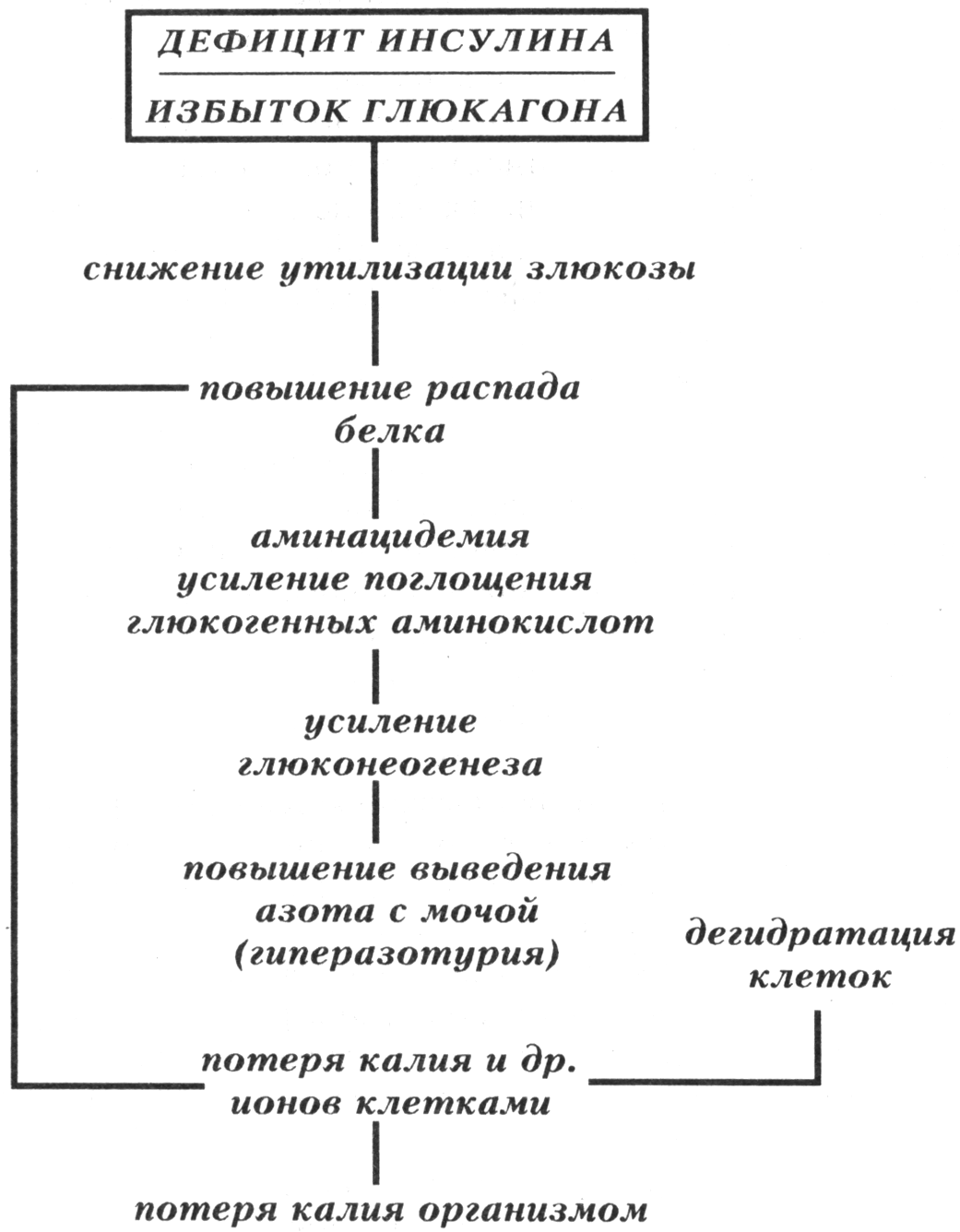
Рис. 14.12. Белковый обмен при сахарном диабете. Пояснения в тексте [по А.Ш. Зайчику, Л.П. Чурилову].
При инсулинорезистентности развивается метаболический Х-синдром, который включает тканевую инсулинорезистентность (в основном мышцы) с компенсаторной гипергликемией, дислипопротеинемия с накоплением атерогенных липидов, артериальную гипертонию и ожирение. Первична инсулинорезистентность. Синдром играет ведущую (или важнейшую) роль в патогенезе сахарного диабета II типа, гипертонической болезни, атеросклероза, ИБС и ИБМ.
Дислипопротеинемия связана с влиянием инсулина на обмен липидов и приводит к увеличению в крови ХС ЛПОНП (реже общего ХС и ХС ЛПНП) и понижение уровня ХС ЛПНП. Это приводит к росту атерогенности крови и ускорению развития атеросклероза.
Повышение АД при инсулинорезистентности связано с эффектами инсулина: активацией симпато-адреналовой системы, увеличение почечной реабсорбции натрия и воды (особенно при сочетании с ожирением), стимуляция факторов клеточного роста (миокард, сосудистая стенка), нарушение трансмембранных ионных механизмов (внутриклеточное накопление натрия и кальция и уменьшение К с усилением прессорных эффектов норадреналина и ангиотензина)
Диабетическая кетоацидотическая кома - является следствием абсолютной или относительной инсулиновой недостаточности и резкого снижения утилизации глюкозы тканями организма. Чаще всего она развивается у больных ИЗД, характеризующимся, как правило, тяжелым лабильным течением.
Содержание глюкагона в сыворотке крови при диабетическом кетоацидозе повышено и наблюдается тесная связь между повышением в крови уровня глюкагона и увеличением концентрации глюкозы и кетокислот. Контринсулярные гормоны, в особенности глюкагон, повышают глюконеогенез, гликогенолиз, липолиз и продукцию в печени кетоновых тел.
Липолиз усиливается настолько, что концентрация триглицеридов, фосфолипидов, холестерина и НЭЖК нередко увеличивается на 50%. Массивное поступление липидов в печень сопровождается ее жировой инфильтрацией и увеличением размеров. Жирные кислоты являются источником энергии преимущественно для мышц, а кетоновые тела – для мозга. Липолиз и протеолиз стимулируются глюкокортикоидами, катехоламинами, СТГ и глюкагоном.
Жирные кислоты метаболизируются в печени до ацетил-КоА, который полностью окисляется до СО2 и Н2О к цикле Кребса или используется для синтеза жирных кислот в процессах липогенеза. При нормальных условиях только небольшая его часть превращается в ацетоацетил-КоА, ацетоуксусную кислоту и в b-гидроксимасляную кислоту. Ацетоуксусная кислота в свою очередь превращается в ацетон. При диабете и особенно при кетоацидотической коме усиленный распад жиров и повышение их поступления в печень приводят к образованию избытка ацетил-КоА и усиленному кетогенезу.
Развивается и аминокислотный дисбаланс, так как в плазме крови повышается концентрация кетогенных аминокислот (лейцин, изолейцин, валин) при относительном снижении уровня глюкогенных аминокислот (глицин, серин, аланин, глутамин), котрые расходуются на глюконеогенез.
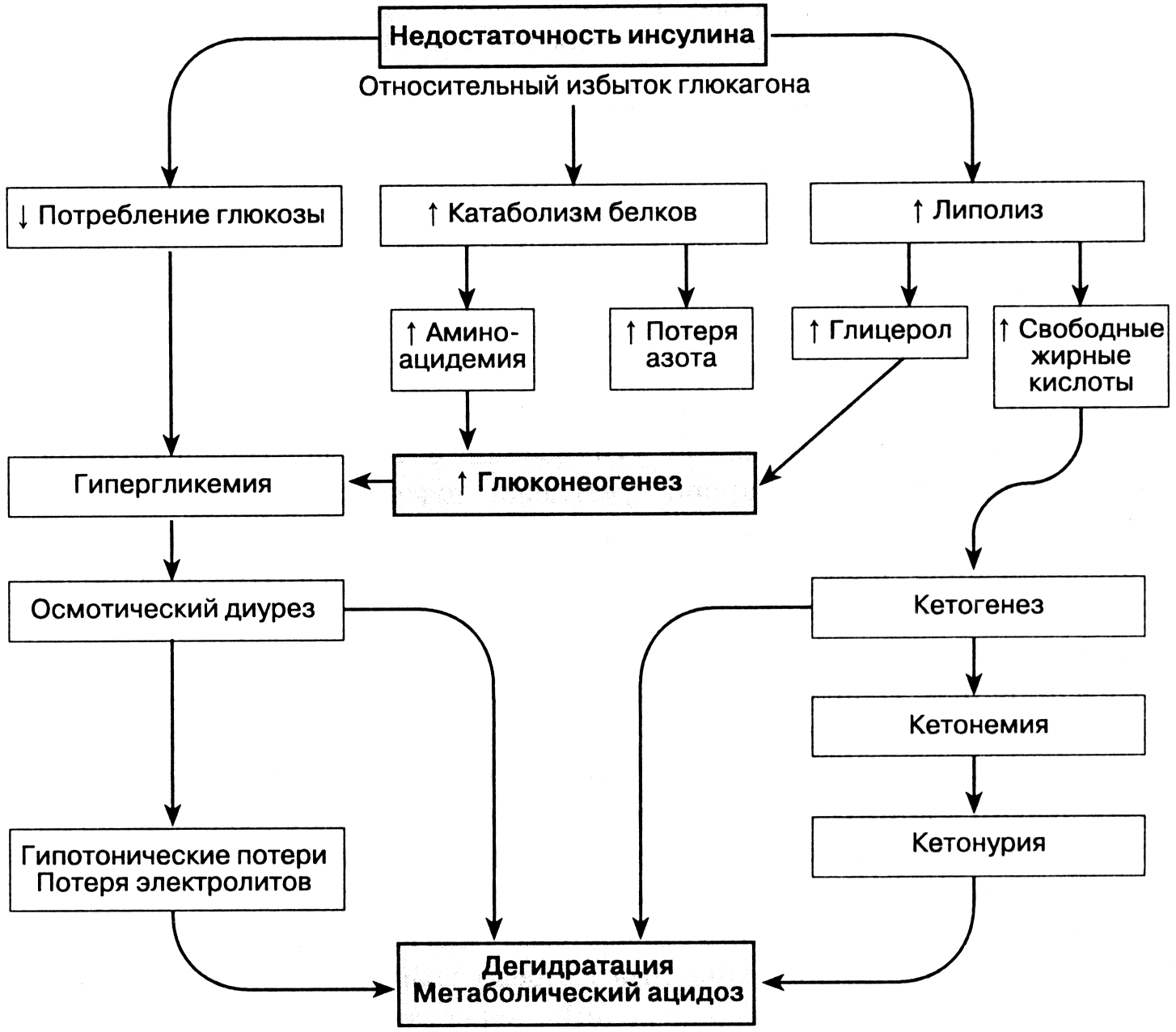
Рис. 14.13. Патогенез диабетического кетоацидоза (ДКА). Диабетический кетоацидоз начинается с недостаточности инсулина. Увеличение концентрации глюкозы, происходящее в результате этого, вместе с усилением катаболизма белков и липолиза ведут к дегидратирующему диурезу, истощению электролитов, тяжелой гипергликемии и выраженному ацидозу. Для коррекции этого состояния необходима заместительная терапия инсулином, электролитами и жидкостью [по В.М. Кэттайл, Р.А. Арки, 2001].
Массивное поступление в печень кетогенных субстратов (липидов и аминокислот) способствуют нарастанию кетогенеза при декомпенсации диабета. Прогрессирующее накопление в крови кетоновых кислот вызывает истощение ее щелочных резервов, в результате чего происходит снижение рН крови и развивается метаболический ацидоз, т.е. кетоацидоз.
Патогенез расстройств сознания и других психоневрологических симптомов диабетической комы до конца не ясен. Эти нарушения принято связывать со следующими факторами:
1) токсическим действием на мозг избытка кетоновых тел;
2) ацидозом ликвора спинномозговой жидкости (возможно, внутриклеточным ацидозом в ЦНС);
3) дегидратацией клеток мозга;
4) гиперосмолярностью внутриклеточного пространства в ЦНС;
5) гипоксией ЦНС вследствие снижения 2,3-дифосфоглицерофосфата;
6) снижением содержания ГАМК в ЦНС.
Гиперосмолярная (некетонемическая) диабетическая кома встречается чаще у лиц, страдающих ИНЗД легкой или средней тяжести, получающих только диетотерапию или сахароснижающие пероральные препараты. Способствуют развитию гиперосмолярной комы различные инфекции (пневмония, пиелит, цистит и др.), острый панкреатит, ожоги, инфаркт миокарда, охлаждение, невозможность утолить жажду (у одиноких престарелых больных, прикованных к постели).
Отсутствие кетоацидоза при гиперосмолярной коме связано с тем, что, с одной стороны, при данной патологии выявляется остаточная секреция инсулина, которая недостаточна для того, чтобы ликвидировать гипергликемию, но вполне достаточна, чтобы ингибировать липолиз, а с другой – чувствительность к инсулину периферических тканей отличается от таковой при кетоацидозе.
Пусковым моментом водно-электролитных нарушений при этом виде комы является гипергликемия, сопровождающаяся повышением осмотического давления в сосудистом русле. Для сохранения изоосмолярности сред начинается компенсаторное перемещение жидкости из клеток и внеклеточного пространства в сосудистое русло. Одновременно развивается глюкозурия и как следствие ее – полиурия, так как высокое осмотическое давление первичной мочи препятствует обратному всасыванию воды в почечных канальцах. Этот так называемый осмотический диурез ведет к массивной потере не только воды, но и электролитов, прежде всего ионов Na+, K+, CI+, HCO3.
Восстановление изоосмолярности между тремя пространствами происходит не за счет снижения гиперосмолярности в сосудистом русле, а за счет клеточной дегидратации и повышения внутриклеточного осмотического давления.
В сравнении с кетоацидозом при гиперосмолярной коме отмечается повышенная склонность к различным гемокоагуляционным нарушениям, особенно к таким, как диссеминированная внутрисосудистая коагуляция (ДВС-синдром), артериальные и венозные тромбозы.
Лактатацидотическая кома встречается при диабете значительно реже, чем кетоацидоз и гиперосмолярная кома. Лактатацидоз может сочетаться с кетоацидозом или гиперосмолярной комой. Иногда он развивается на фоне приема бигуанидов (фенформина и адебита) у больных с сердечной и почечной недостаточностью, заболеваниями печени, легких, а также при шоке, кровопотере, сепсисе.
Известно, что печень способна метаболизировать около 3400 ммоль молочной кислоты в сутки. Но при перечисленных выше состояниях, сопровождающихся значительной тканевой гипоксией, образование молочной кислоты превалирует над процессами ее утилизации. Развитию лактатацидоза способствует также парентеральное введение жидкостей, содержащих фруктозу, сорбит или ксилит.
Общая схема лечения диабетический комы включает:
ликвидацию инсулиновой недостаточности и нормализацию углеводного обмена;
оптимально быструю регидратацию организма;
восстановление нормального вне- и внутриклеточного электролитного состава;
восстановление запасов глюкозы (гликогена) в организме;
восстановление нормального кислотно-основного равновесия (КОР);
диагностику и лечение заболеваний или патологических состояний, вызвавших диабетическую кому;
проведение комплекса лечебных мероприятий, направленных на восстановление и поддержание функций внутренних органов (сердца, почек, легких и др.).
Гипогликемическая (инсулиновая) кома развивается у больных СД при введении избыточной дозы инсулина или сульфаниламидов (особенно в сочетании с салицилатами, алкоголем) на фоне недостаточного потребления углеводов с пищей или при наличии инсулиномы.
Гипогликемическую кому, безусловно, определяет нейрогликопения (снижением уровня глюкозы в головном мозге). В основе патогенеза гипогликемической гипоксии лежит энергетическое голодание нейронов, дезорганизация окислительно-восстановительных процессов в нейронах, т.е. острая субстратная гипоксия мозга
Именно клетки головного мозга наиболее чувствительны к гипогликемии, поскольку они получают энергию за счет аэробного окисления глюкозы и не способны:
1) накапливать глюкозу в значительных количествах;
2) синтезировать глюкозу;
3) метаболизировать другие субстраты, кроме глюкозы и кетоновых тел. Причем, последние удовлетворяют энергетические потребности мозга в незначительной степени;
4) извлекают в недостаточных количествах глюкозу из внеклеточной жидкости; инсулин не способствует поступлению глюкозы из внеклеточной жидкости в нейроны.
Гипогликемические комы, связанные с передозировкой инсулина, требуют коррекции глюкозой, а для их предотвращения и подробной инструкции больному насчет времени введения препарата относительно приема пищи.
41. – Нарушение переваривания и всасывания липидов. Гипо-, гипер- и дислипопротеинэмии. Атерогнность крови. Жировая инфильтрацияорганов.
Нарушение переваривания и всасывания
Основной симптом – стеаторея (появление липидов в кале).
Причины: дефицит панкреатической липазы (панкреатическая), снижение секреции желчи (гепатогенная), угнетение ресинтеза триглицеридов в кишечнике при его заболеваниях (энтерогенная).
При нарушении переваривания жиров не формируются смешанные мицеллы нормальной структуры, в составе которых всасываются продукты гидролиза жиров, холестерин, жирорастворимые витамины, полиеновые эссенциальные жирные кислоты.
Стеаторея может привести к уменьшению всасывания кальция, т.к. жирные кислоты образуют с ионами Ca++ нерастворимые соли, которые плохо всасываются.
Нарушение переваривания и всасывания липидов в кишечнике обычно коррегируют назначением двух групп препаратов ‑ препаратов желчных кислот для эмульгирования и ферментных препаратов, обычно комплексных, но обязательно включающих и липазы. Вместо заместительной терапии можно применить и желчегонные средства ‑ препараты, повышающие секрецию желчи и препараты, способствующие отделению желчи. Поскольку основным поставщиком липазы является поджелудочная железа, применяют и вещества, повышающие ее функцию (прямо или косвенно повышающие тонус блуждающего нерва) ‑ стимуляторы центра блуждающего нерва, М-холиномиметические препараты или антихолинэстеразные.
Поскольку следствием стеатореи являются авитаминозы (А, Д, Е, К), требуется соответствующая заместительная терапия витаминами.
Гипо-, гипер- и дислипопротеинемии. Атерогенность крови. Фармакологическая коррекция дислипопротеидемий.
Гиперлипидемия - основное проявление нарушения процессов транспорта жиров в крови и их перехода в ткани.
Основные виды гиперлипидемий:
Алиментарная - избыточное употребление жиров животного происхождения, несбалансированный рацион (много жиров и мало липотропных факторов).
Транспортная - при обеднении печени гликогеном и при голодании усиливается распад жиров и поступление их в кровь (голодание, стрессовые ситуации).
Ретенционная (эндогенная) - задержка перехода жиров из крови в ткани при:
низкой наследственной активности липопротеинлипазы,
нарушении активации липопротеинлипазы или образовании ее ингибиторов (атеросклерозе, постгеморрагических состояниях, облучении, сахарном диабете, механической желтухе, избытке NaCl, в пожилом возрасте),
дефиците альбуминов, участвующих в транспорте жиров (нефроз, гепатит, при стрессе).
К основным последствиям гиперлипидемии относятся: ожирение, жировая инфильтрация и дистрофия печени, холестериноз, претромботические состояния, атеросклероз, ИБС, инсульт.
Гиперлипопротеидемия (ГЛП) – повышение одного или нескольких классов липопротеидов.
Гиполипопротеидемия – снижение одного, реже двух классов липопротеидов.
Таблица 15.4.
Основные виды гиполипопротеидемий (ГЛП)
|
Гипо--липопротеидемия |
А--липопротеидемия |
Гипо-α-липопротеидемия |
А-α-липопротеидемия |
Сочетание гипер- α-липопротеидемии с гипо--липопротеидемией |
Прозрачность плазмы |
Прозрачная |
Прозрачная |
Прозрачная |
Прозрачная или слегка мутная |
Прозрачная |
ХС общий |
|
|
N или |
|
N или |
ТГ |
|
|
N |
|
|
ХС ЛПНП |
|
или 0 |
N |
N |
|
ХС ЛПВП |
N |
N |
|
|
|
Хиломикроны
|
Отсутствуют |
Отсутствуют |
N |
N |
N |
Пре--ЛП |
|
Отсутствуют |
N |
Появление патологических ЛПОН |
или 0 |
-ЛП |
|
Отсутствуют |
N |
N |
|
α-ЛП |
N |
N |
|
|
|
Клиническая картина |
Симптомы поражения нервной и мышечной систем (атаксия, потеря сухожильных рефлексов); поражение сетчатки. |
Ранний атеросклероз |
Неврологические расстройства, спленомегалия, увеличение лимфоузлов и миндалин (желтого цвета) |
|
|
Примечание. - снижено; - повышено; - резко снижено; - резко повышено; N – норма.
Дислипопротеидемия – повышение, снижение или отсутствие в крови одного или нескольких классов липопротеидов.
Первичные гиперлипопротеинемии связаны с генетическими нарушениями (например, с недостатком рецепторов ЛПНП).
Вторичные гиперлипопротеинемии развиваются при ряде заболеваний (сахарный диабет, нефротический синдром, гипотиреоз и др.) или при применении лекарственных средств, которые повышают уровни атерогенных липопротеинов (тиазидные диуретики, -адреноблокаторы, пероральные контрацептивные средства и др.)
Таблица 15.5.
Типы гиперлипопротеинемий и их возможные причины.
Фенотип |
Уровень каких ЛП повышен |
Причина |
Первичные причины |
Вторичные причины |
I |
Хиломикроны |
Дефицит липопротеинлипазы, нарушение гидролиза ХМ |
Дефицит липопротеинлипазы, дефицит апо С-II |
Системная красная волчанка (редко) |
IIа |
ЛПНП |
Замедление распада ЛПНП |
Семейная гиперхолистеринемия |
Пониженная функция щитовидной железы |
IIб |
ЛПНП и ЛПОНП |
Происхождение неизвестно |
Комбинированная семейная гиперхолистеринемия |
Диабет, нефротический синдром, анорексия |
III |
Ремнанты хиломикронов и ЛППП |
Замедление распада ЛПОНП (пре-) и А-пре- с апопротеином А |
Семейная гиперлипопротеинемия III типа |
Пониженная функция щитовидной железы, диабет, ожирение |
IV |
ЛПОНП |
Следствие гиперинсулинизма |
Комбинированная гиперлипидемия, семейная гипертриглицеридемия |
Диабет, хронические заболевания почек |
V |
Хиломикроны и ЛПОНП |
Происхождение неизвестно |
Семейная гипертриглицеридемия, дефицит апо С-II |
Алкоголь, -блокаторы, диуретики, гормональные контрацептивы |
Наиболее атерогенны II, III, IV типы гиперлипопротеинемий.
Таблица 15.6.
Изменение содержания холестерола, триглицеридов и липопротеинов плазмы при различных видах гиперлипопротеинемий
Изменение содержания |
Тип нарушения |
|||||
I |
II а |
II б |
III |
IV |
V |
|
Холестерола |
0, + |
+ |
+ |
++ |
0, + |
+ |
Триглицеридов |
+++ |
0 |
++ |
+++ |
+++ |
++ |
Хиломикронов |
++ |
0 |
0 |
0 |
0 |
++ |
-липопротеинов |
- |
0 |
0 |
0 |
0 |
0 |
-липопротеинов |
- |
++ |
++ |
А--ЛП |
0 |
0 |
Пре--липопротеинов |
- |
0 |
+ |
А-пре--ЛП |
++ |
++ |
Примечание. А--ЛП и А-пре--ЛП - аномальные бета- и пребеталипопротеины; "0", "-", "+" обозначают отсутствие изменений, уменьшение и увеличение; число знаков "+" - примерная степень изменения (малая, средняя и большая).
Для оценки атерогенности плазмы крови предложен коэффициент атерогенности Климова (КА), который в норме должен быть ниже 3,5 у.е.:
ХС - ХС ЛПВП КА = -------------------------- ХС ЛПВП |
Нарушения метаболизма холестерина [по Ю.М. Лопухину и соавт., 1983]
Тип I. Изменение содержания ХС в организме.
Холестериноз (избыточное отложение холестерина в клетках).
а) неосложненный (старение в связи с ослаблением стероидогенеза);
б) осложненный (атеросклероз).
Холестериновые дефициты.
а) злокачественные новообразования;
б) вирусные инфекции.
Тип II. Изменение содержания ХС в плазме крови.
Первичные (наследственные) гиперхолестеринемии.
а) семейные (дефект рецепторов ЛПНП);
б) семейные III типа обусловленные нарушением желчегенеза.
Вторичные (приобретенные) гиперхолестеринемии.
а) механическая желтуха;
б) билиарный цирроз печени;
в) сахарный диабет;
г) гипотиреоз;
д) нефроз;
е) гиперглобулинемия (системная красная волчанка и др.);
ж) синдром Кушинга;
з) гематома;
и) беременность.
Тип III. Накопление ХС в отдельных органах и тканях (ксантоматоз, ожирение).
Жировая инфильтрация - избыточное отложение жиров в тканях, не относящихся к жировой (чаще печень).
Виды жировой инфильтрации:
Алиментарная.
Эндогенная.
Алипотропная.
Анизотропная.
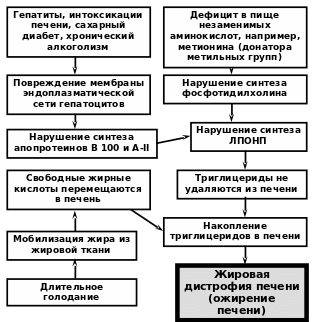
Рис. 15.10. Схема патогенеза жировой инфильтрации печени.
Положительный эффект при жировой инфильтрации печени дает введение донаторов метильных групп: метионин, В15, В12, холин.
42. – Ожирение и его виды. Патогенез алиментарного ожирения. Виды и особенности патогенеза вторичного ожирения. Принципы патогенетической терапии ожирения.
Ожирение – это увеличение отложения жира в жировых клетках (адипоцитах).
Классификация ожирения
По этиологии
Первичное (алиментарно-конституциональное или экзогенно-конституциональное).
Вторичное (симптоматическое).
Церебральное ожирение
опухоли головного мозга
травма основания черепа и последствия хирургических операций
синдром пустого турецкого седла
травмы черепа
воспалительные заболевания (энцефалит и др.)
Эндокринное ожирение
гипофизарное
гипотиреоидное
климактерическое
надпочечниковое
смешанное
Ожирение на фоне психических заболеваний и/или приема нейролептиков
По типу отложения жира
Генерализованное и местное.
Андроидное (по мужскому типу).
Гиноидное (по женскому типу).
По характеру жировой ткани
Гипертрофическое (увеличение размера жировых клеток).
Гиперпластическое (увеличение количества жировых клеток).
Смешанное.
По степени (нормальная масса тела рассчитывается разными способами по индексу Брока = (рост, см – 100) 10%; индексу Кетле – рассчитывается как отношение массы тела к росту возведенному в квадрат, в норме он не должен превышать 25; по таблицам).
Степень ожирения рассчитывается как отношение должной массы тела к измеренной и умноженное на 100%:
15 - 29%
30 - 49%
50 - 99%
100% И выше
Рис. 15.6. Отложение жира при андроидном и гиноидном типах ожирения [по Я. Татонь, 1981].
Первичное ожирение (или алиментарно-конституциональное) развивается при избытке поступающей в организм с пищей энергии по сравнению с ее расходом.
Патогенез первичного ожирения
Алиментарный дисбаланс (избыточная энергетическая ценность питания) – ведущий фактор первичного ожирения.
Контроль за отложением энергетических субстратов - жиров (триацилглицеринов - ТАГ) в адипоцитах и их мобилизацией происходит с участием гормонов. После еды при увеличении концентрации глюкозы в крови синтез жиров под влиянием инсулина в адипоцитах увеличивается.
Инсулин в адипоцитах активирует следующие процессы:
транспорт глюкозы в клетки;
реакции гликолиза и пентозофосфатного пути, необходимые для синтеза жирных кислот и α-глицерофосфата;
поступление жирных кислот в клетки под действием ЛП-липазы и их синтез из продуктов распада глюкозы;
синтез жиров;
ингибируется липолиз (в результате дефосфорилирования гормончувствительной липазы при участии инсулина).
Поэтому отложению жира способствует прием пищи, богатой жирами и углеводами.
При первичном ожирении возможен функциональный гиперинсулинизм.
Нарушение регуляции чувства голода и насыщения
Количество потребляемой пищи зависит от регуляции чувства голода и насыщения, которые определяются концентрацией в крови глюкозы и гормонов пищеварительного тракта, которые инициируют чувство насыщения (холецистокинин-панкреозимин, нейротензин, бомбезин). Выделяясь в кровь при поступлении пищи в пищеварительный тракт, они возбуждают центр насыщения (вентро-медиальные ядра гипоталамуса) и тормозят центр голода (латеральные ядра гипоталамуса). Аналогичные эффект вызывает растяжение желудка пищей и возбуждение его рецепторов вазоактивным интерстициальным полипептидом, холецистокинином-панкреозимином.
При первичном ожирении нарушается выработка данных веществ, а желудок растянут (имеет большой объем), поэтому требуется значительный объем пищи для возбуждения его рецепторов растяжения. Может страдать периферический механизм (снижение чувствительности рецепторов желудка и гипоталамуса к гормонам пищеварительного тракта).
Генетические различия метаболизма у тучных и худых людей.
Генетически детерминированная разница в функционировании бесполезных циклов: эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идет с затратой АТФ. Если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит «бесполезный» расход АТФ и, соответственно, источников энергии, например жиров.
У людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм.
У людей возможно разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз как менее эффективный «сжигает» гораздо больше глюкозы, в результате снижается ее переработка в жиры.
Наследственность
Ген ожирения (obese gene) обнаружен у человека и других млекопитающих. Одиночные мутации в этом гене приводят к избыточной массе тела. Продуктом экспрессии гена ob является белок ob (или «лептин», от греч. leptos - тонкий), он секретируется в кровь адипоцитами. Одним из органов-мишеней белка ob является центральная нервная система (ЦНС), через которую гормон осуществляет свое действие. Белок ob увеличивает энергетический обмен, потребление кислорода, температуру тела, двигательную активность, снижает потребление пищи уменьшается масса тела вследствие снижения количества жиров в организме.
Нарушение чувствительности к липолитическим гормонам: адреналин, глюкагон, СТГ, тироксин.
Психологический фактор – удовольствие от приема пищи, обусловленное выработкой эндорфинов и энкефалинов.
Культурный фактор – национальные и семейные традиции питания, например, пища, богатая жирами в северных районах, привычки «заедать» просмотр кинофильма, чтение книг, работу на компьютере.
Теории ожирения
Глюкостатическая теория – повышенное потребление пищи обусловлено снижением чувствительности глюкорецепторов центра насыщения.
Липостатическая теория - повышенное потребление пищи обусловлено повышением в крови концентрации жирных кислот и стимуляции ими центра голода. Концентрация ЖК в плазме повышается при голодании (мобилизация ЖК из адипоцитов), а также при приеме пищи, богатой жирами.
Патогенетическая терапия первичного ожирения
Первичное ожирение, прежде всего, лечится препаратами, понижающими аппетит (анорексики). В основном, это препараты, восполняющие дефицит адренорецепции на уровне стимуляции центра насыщения и последующего угнетения им центра голода (мазиндол, фенамин, дезопимон). Данная группа препаратов ‑ центральных адреномиметиков обычно не лишена и периферического подобного действия, за счет чего из-за усиленного гликогенолиза возникает гипергликемия, которая сама по себе снижает аппетит. Правда, гипергликемия и последующая секреция инсулина несколько усилят липогенез, однако последнее компенсируется адреномиметическим липолитическим действием упомянутых препаратов на уровне адипоцита.
Из этих же соображений при ожирении показано назначение других препаратов, усиливающих “сжигание жира”, активаторов липолиза - веществ, усиливающих метаболические процессы.
Другой подход к снижению аппетита – угнетение центра голода за счет высвобождения серотонина в центральной нервной системе (фенфлурамин), кроме того, данная группа препаратов усиливает и липолиз.
Кроме этого применяются слабительные и набухающие средства (целлюлоза, агар).
Рис. 15.7. Схематическое изображение систем, контролирующих постоянство массы тела и механизмов первичного ожирении. Сокращения: ГПП- I - глюкагоноподобныи пептид I, CHC - симпатическая нервная система, ЛПГ – липотропный гормон [по А.Ш. Зайчику, Л.П. Чурилову, 2000].
Вторичное ожирение (или симптоматическое ожирение) - это тип ожирения, которое развивается в результате какого-то основного заболевания (чаще всего эндокринного).
Гипофизарное ожирение (болезнь Иценко-Кушинга, центральный гиперкортицизм) развивается в результате гиперпродукции гипофизом (чаще всего вследствие базофильной аденомы гипофиза) адренокортикотропного гормона (АКТГ) стимулирует выработку глюкокортикоидов (в пучковой зоне коры надпочечников) и половых гормонов (в сетчатой зоне коры надпочечников).
Характерно лунообразное багрово-красное лицо, гипертрихоз, стрии, увеличение АД, гипергликемия.
Ожирение носит диспластических характер (верхняя половина туловища и лицо при худых конечностях). Глюкокортикоиды вызывают гипергликемию избыток глюкозы утилизируется преимущественно инсулинзависимыми тканями (например, жировой), особенно теми, в которых много рецепторов к инсулину (верхняя половина туловища и лицо) избыток поступившей в адипоциты глюкозы превращается в триглцериды и откладывается.
Надпочечниковое ожирение (синдром Иценко-Кушинга, первично-гландулярная форма гиперкортицизма) развивается при повышенной продукции глюкокортикоидов (вследствие опухоли пучковой зоны коры надпочечников) АКТГ снижается по принципу обратной связи. Наблюдается одностороннее увеличение одного надпочечника при гипотрофии другого.
Гипотиреоидное ожирение - следствие недостаточности функции щитовидной железы синтезировать тироксин и трийодтиронин (нет или значительно снижен катаболический эффект тиреоидных гормонов).
Ожирение при опухоли -клеток островкового аппарата поджелудочной железы (секретирующая инсулинома) – развивается вследствие избытка инсулина, который тормозит липолиз и стимулирует липогенез. Характерна гипогликемия.
Гипоовариальное и климактерическое ожирение - результат снижения секреции половых гормонов → снижение основного обмена преобладание анаболических процессов. Снижение секреции гонадотропинов (обладающих липотропным действием) → снижение процессов липолиза.
Ожирение при адипозогенитальной дистрофии - заболевание, связанное с поражением гипоталамо-гипофизарной системы (внутриутробная инфекция (токсоплазмоз), перинатальная травма, инфекции (скарлатина, вирусные инфекции, туберкулез) и травматические поражения мозга, опухоли (краниофарингиома, хромофобная аденома), тромбозы, эмболии, кровоизлияния) и характеризующееся недоразвитием половых желез и ожирением.
При поражении гипоталамуса происходит повреждение или раздражение его паравентрикулярных и вентромедиальных ядер, что ведет к резкому повышению аппетита с последующим развитием ожирения. Вследствие поражения гипоталамуса снижается гонадотропная функция гипофиза. Это в свою очередь приводит к гипогонадизму с последующим изменением высшей нервной деятельности и развитием ожирения.
Рис 15.8. Нарушение регуляторных механизмов при адипозогенитальной дистрофии [по В.В. Потемкину, 1986].
Патогенетическая терапия вторичного ожирения
Вторичные ожирения при гипотиреозе лечатся аналогами тироксина, ожирения при гипогонадизмах (гипоовариальное, климактерическое, евнухоидное) ‑ коррекцией соответствующими полу половыми гормонами (эстрогенами или андрогенами); кушингоидные ‑ снижением уровня секреции глюкокортикоидов (метирапон, митотан), или уменьшением их периферического действия антагонистами на уровне рецепторов (мифепристон).
Фармакотерапия жирового истощения зависит от причины, ее вызвавшей: психогенная анорексия требует коррекции функции нервной системы седативными и антидепрессантами, тиреотоксикоз ‑ антитиреоидными средствами и т. д. Но в любом случае для усиления липогенеза будет эффективна терапия глюкозой и инсулином ‑ пока единственным доступным гормональным препаратом, усиливающим липогенез. Ранее предпринимались попытки увеличить количество жиров путем блокады липолиза ингибиторами ферментов (довольно токсичными препаратами мышьяка), однако, в настоящее время они оставлены из-за возможных побочных эффектов. С целью повышения аппетита издавна применяют рефлекторную стимуляцию центра голода с рецепторов ротовой полости (горечи) или путем снижения уровня глюкозы в крови (инсулином).
Нарушения метаболизма у людей, страдающих ожирением
У людей с избыточным количеством жировой ткани имеются следующие общие отклонения от нормального обмена веществ:
снижена толерантность к глюкозе (может восстанавливаться при снижении массы тела);
гипертриглицеридемия в результате гиперлипопротеинемий II-V типов, чаще всего повышена концентрация ЛПОНП и ЛПНП;
гиперинсулинемия;
снижена продукции гормона роста.
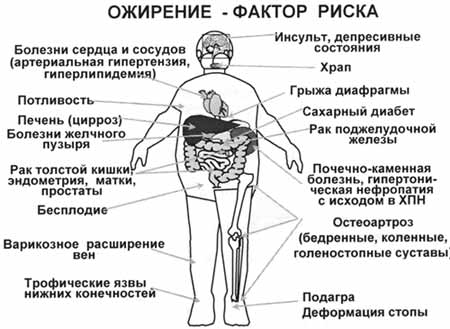
Рис. 15.9. Ожирение как универсальный фактор риска развития различных заболеваний [http://www.endocrinolog.ru/illnesses/obesity/obesity_risk.htm].
43. – Атеросклероз. Этиология. Факторы риска и меры профилактики. Стадии развития атеромы.
Атеросклероз («athere» - кашицеобразная масса (греч.), «sclerosis» – твердый) - хроническое заболевание, характеризующееся поражением стенки артериальных сосудов, в основе которого лежит нарушение липидного обмена, отложение в сосудистой стенке жировых масс (ХС) с утолщением и деформацией стенки.
Основная причина смерти: ишемическая болезнь сердца (60% мужчин и 41% женщин), ишемическая болезнь мозга (25% мужчин и 39% женщин, 1991).
Стадии атеросклероза
Долипидная стадия - создаются условия для проникновения липидов в интиму, обнаруживаются очаговые микроповреждения эндотелия (повышения проницаемости), изменения интерстициальной ткани (расширение межклеточных каналов), клеточных структур (набухание), эластолиз (деструкция эластических мембран с накоплением холестерина). Все это способствует в дальнейшем липоидной инфильтрации внутренней поверхности артерий. Продолжительность стадии зависит от способности протеолитических и липолитических ферментов, находящихся в интиме, растворять и выводить продукты нарушенного обмена.
Стадия липоидоза (жировые пятна и полоски) начинается с накопления в интиме ЛПНП в комплексе с иммуноглобулинами, а также фибрина, образования комплексов атерогенных липопротеинов с глюкозаминогликанами интерстициальной ткани, что сочетается с изменением аминокислотного состава эластина и ведет к набуханию эластических волокон, фрагментации внутренней эластической мембраны, разволокнению и набуханию внутренней оболочки сосудов. При этом активизируются гладкомышечные клетки интимы и макрофаги, которые начинают поглощать липиды и трансформироваться в ксантомные (пенистые) клетки.
Стадия липосклероза проявляется разрастанием молодой соединительной ткани в участках отложения липопротеидов, созревание соединительной ткани ведет к образованию фиброзных бляшек. Вследствие нарушений эндотелия на поверхности интимы в местах скопления липидов может произойти агрегация тромбоцитов или выпадение нитей фибрина, что, вместе взятое, является началом фибропластического процесса. Дальнейшее прогрессирующее накопление липидов может быть обусловлено тем, что причины, его вызвавшие, не смогли быть устранены и продолжают оказывать свое действие. В результате этого изменяется метаболизм гладкомышечных клеток сосудов, и они теряют способность утилизировать поступающие жиры, которые по мере гибели клеток попадают в окружающие ткани, где они располагаются в основном веществе и вдоль эластических волокон. Высвободившиеся липиды, фибриноген пропитывают бляшку, а выпавший фибрин оказывает фибропластическое, склерозирующее действие. Возникает типичная фиброзная бляшка, состоящая из бедной клеточными элементами грубой соединительной ткани, эластических волокон и того или иного количества жировых масс с кристаллами холестерина в центре.
|
Рис. 15.11. Стадия липоидоза, макропрепарат аорты (жировые пятна и полоски показаны стрелками) [http://www.its.caltech.edu/~sciwrite/journal03/roth.html, Diehm, 2003]. |
Рис. 15.12. Строение типичной фиброзной атеросклеротической бляшки [http://wcb.ucr.edu/wcb/schools/CNAS/bmsc/lloo/10/modules/page9.html]. 1 - некротизированный центр: клеточный детрит, кристаллы холестерина, пенистые клетки, кальций; 2 – фиброзная капсула: гладкомышечные клетки, макрофаги, пенистые клетки, лимфоциты, коллаген, эластин, протеогликаны; 3 – медиа.
Стадия атероматоза (осложненная бляшка) характеризуется распадом в зоне бляшки липоидов, коллагеновых волокон, а также мышечных и ксантомных клеток. В результате образуется полость, содержащая жиро-белковый детрит (атероматозные массы) и отделенная от просвета сосуда прослойкой соединительной ткани, которая играет роль покрышки бляшки. Прогрессирование атероматоза приводит к осложненным поражениям сосудов - кровоизлияниям в бляшку, разрушению ее покрышки и изъязвлению. Выпадающий при этом в просвет сосудов детрит может стать источником эмболии, а сама атероматозная язва - служить основой для образования тромбов.
Рис. 15.13. Атеросклеротическая бляшка в просвете сосуда в стадии атероматоза [http://www.brown.edu/Courses/Digital_Path/Heart/atherosclerosis.htm].
Стадия атерокальциноза - отложение солей кальция в атероматозные массы, межуточное вещество и фиброзную ткань (петрификация).
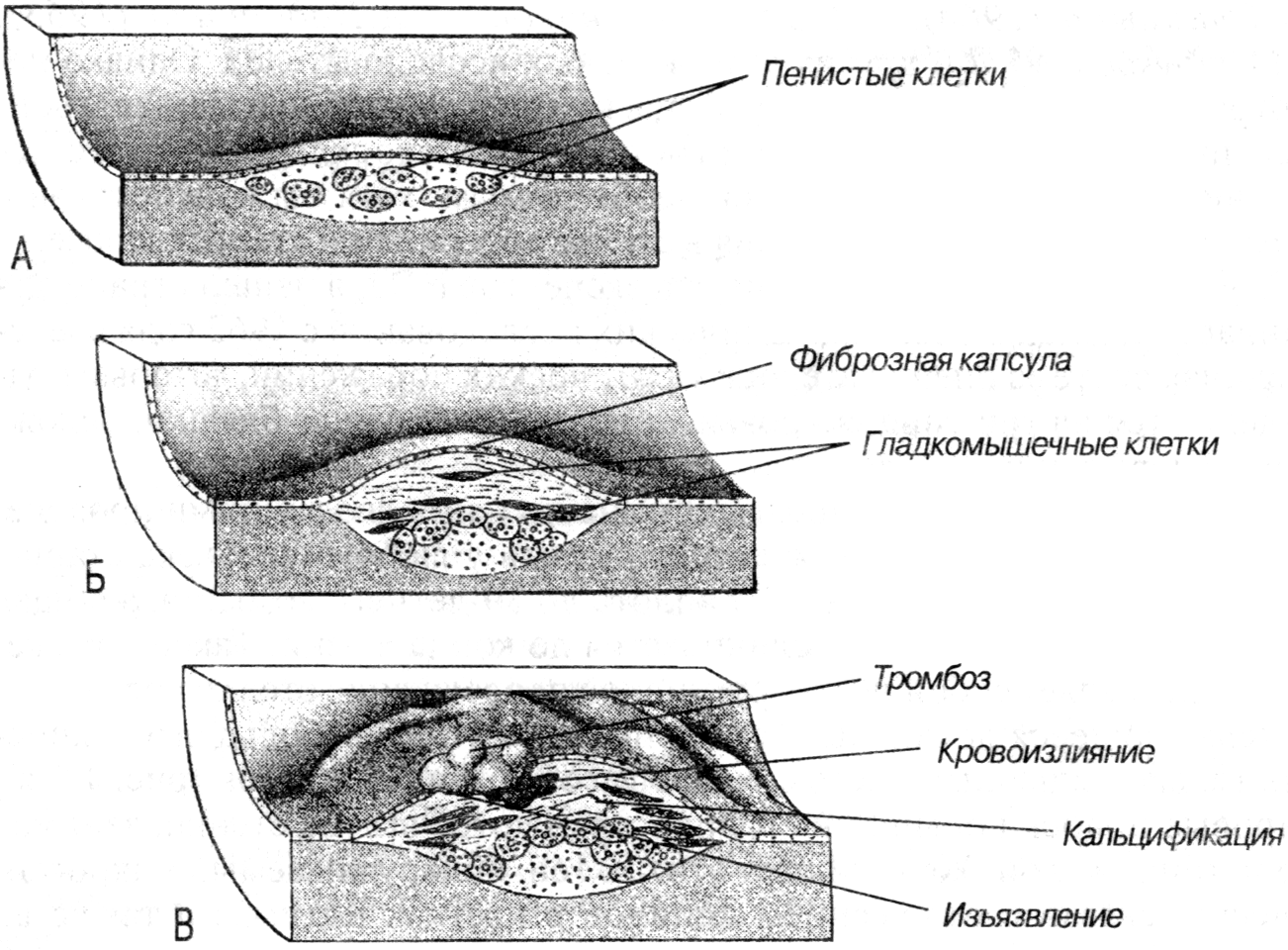
Рис. 15.14. Стадии атеросклероза: жировая полоска (А), фиброзная бляшка (Б), осложненное поражение (В) [по Р.С. Карпову, В.А. Дудко, 1998].
Атеромы локализуются в коронарных, подвздошных, бедренных, сонных, мозговых артериях, аорте, почечных артериях.
Рис. 15.15. Схема гемодинамических нарушений в артериальном сосуде, просвет которого сужен (стенозирован) атеросклеротическими бляшками [http://www.healthandage.com/ html/res/primer/heart.htm].
Атеромы вызывают стенозы (сужение просвета сосуда) и окклюзии (закупорку сосуда) артерий, что приводит к нарушениям гемодинамики в дистальном артериальном русле, ишемии и гипоксии органов и тканей.
При осложненных атеромах развиваются тромбозы и эмболии.
Таблица 15.7.
Факторы, повышающие риск заболеваний, связанных с атеросклерозом [Россия, 1996]
Модифицируемые факторы |
Немодифицируемые факторы |
|
образ жизни |
биохимические и физиологические факторы |
личностные параметры |
Высококалорийное питание с повышенным содержанием животного жира и ХС Курение Избыточное потребление алкоголя Сниженная физическая активность |
Гиперхолестеринемия за счет ЛНП Артериальная гиперто ния Низкий уровень в крови ЛВП Гипертриглицеридемия Ожирение Сахарный диабет Тромбогенные факторы |
Возраст Пол Наличие у близких родственников клинических проявлений атеросклероза (для мужчин - до 55 лет, для женщин — до 65 лет) Наличие в анамнезе семьи больного гиперлипидемий |
Современные факторы профилактики и лечения
Рыбные продукты (содержат жирные -кислоты).
Алкоголь (20-30 г этанола, 50 г водки, 200 г сухого вина, бутылка пива).
Чеснок (алликор, аллисат).
Витамин Е.
Аспирин.
44. – Патогенез атеросклероза. Теории развития атеросклероза. Важнейшие проявления атеросклероза. Методы диагностики.
Этиология и патогенез атеросклероза
Условно все многообразие теорий развития атеросклероза можно разделить на две группы:
Развитие атеросклероза обусловлено факторами, находящимися в плазме крови (липиды, липопротеины, холестерин, фибрин).
Причина атеросклероза - в клеточных, соединительнотканных и других структур артериальной стенки, наступающих под воздействием различных факторов.
В изучении атеросклероза можно выделить 4 этапа фундаментальных исследований, в каждом из которых доминировала идеология лидирующей в тот период научной дисциплины:
Этап патоморфологических исследований, длившийся с конца XIX века до 40-х годов XX века.
Биохимический этап, пик в развитии которого наблюдался в 40-60-х годах XX века.
Этап клеточной биологии, получивший развитие в 70-80-е годы.
Этап молекулярной биологии и клеточной генетики, который является определяющим направлением в настоящее время.
Инфильтрационно-комбинационная (холестериновая) теория атеросклероза Н.И. Аничкова [1915, 1935] базируется на положении, согласно которому основная часть энергетических потребностей артериальной стенки, особенно ее бессосудистых структур (интимы и внутренней трети медии), восполняется за счет липидов плазмы крови. Делается допущение, что плазменные липиды поступают в сосудистую стенку путем просачивания (инфильтрации) плазмы в направлении от эндотелия к адвентиции. Предполагается также, что в норме липиды просачивающейся плазмы проходят без задержки в адвентицию и удаляются через систему лимфатических сосудов. Однако, когда количество липидов велико, они накапливаются в сосудистой стенке, вызывая развитие липидоза..
Весомым подтверждением холестериновой концепции заболевания являются случаи гомозиготной гиперхолестеринемии, при которой тяжелейший атеросклероз развивается в юношеские и даже детские годы, и только снижение уровня холестерина в крови, каким бы путем оно ни достигалось, спасает больных от инфаркта миокарда и неминуемой гибели.
Фибриновая теория Рокитанского [1852] - вследствие отложения фибрина на поверхности артерий и происходит, собственно, образование бляшек и сужение сосуда, т.е. атеросклеротическая бляшка состоит из веществ, циркулирующих в крови, и, в первую очередь, - из фибрина. Рокитанский не придавал значения липидам плазмы крови в патогенезе атеросклероза, и в этом заключается недостаток его относительно справедливой гипотезы.
Тромболипидная теория [J. Duguid,1946; I. Mustard, 1961] - липиды, накопившиеся в артериальной стенке, каким-то образом «притягивают» к себе фибрин, а последний, в свою очередь, обладает способностью захватывать липиды; допускается также возможность заноса липидов тромбоцитами, задерживающимися между нитями фибрина на поверхности сосуда.
Перекиси липидов ингибируют в эндотелиальных клетках артерий фермент простациклин-синтетазу развивается локальная недостаточность простациклина при сравнительно высоком содержании тромбоксана агрегация тромбоцитов на поверхности эндотелия, что способствует развитию атеросклероза.
Теория повреждения эндотелия артериальной стенки - эндотелий играет роль защитного барьера в отношении атеросклероза и не играет активной роли в накоплении липидов и липопротеинов в артериальной стенке; только повреждение эндотелия вследствие воздействия гемодинамических факторов (кровяное давление, турбулентность потока крови, ее боковое давление), токсинов (никотин, некоторые лекарственные препараты), воспалительных процессов (вирусных, бактериальных, иммунологических), образования тромбов способствует проникновению макромолекулярных соединений из плазмы крови в артериальную стенку и только в местах ее повреждения.
Перекисная теория придает значение перекисям липидов, образующимся в результате свободнорадикального окисления ненасыщенных жирных кислот, а также образующейся гидроперекиси холестерина. Предполагается, что проникновение липопротеинов, содержащих окисленные фосфолипидные ацилы и гидроперекиси холестерина, в стенку сосуда или образование перекисей липидов в самой стенке могут вызвать первичное повреждение интимы и усилить течение атеросклеротического процесса.
Моноклональная теория E. Benditt [1974] рассматривает атеросклеротическое поражение как доброкачественную опухоль, вызванную вирусами или химическими веществами окружающей среды, поскольку для атеросклеротических поражений характерна пролиферация гладкомышечных клеток, рост количества коллагена, эластина и самой бляшки в целом.
Мембранная теория R. Jackson и Л. Gotta [1976] объясняют причину пролиферации гладких мышечных клеток.
Поступление в клетку избыточного холестерина снижает жидкостность мембраны для поддержания жидкостного состояния мембраны клетка увеличивает синтез жирных кислот, с помощью которых эстерифицирует избыток холестерина эфиры холестерина, в отличие от неэстерифицированной его формы, не включаются в фосфолипидный бислой мембраны и могут рассматриваться как защитная для клетки форма избыточного холестерина.
Если способность клетки синтезировать жирные кислоты и эстерифицировать холестерин исчерпана, то начинается пролиферация гладкомышечных клеток, чтобы утилизировать избыток холестерина на построение мембран вновь образующихся клеток.
Аутоиммунная теория А.Н. Климова и др. (1980-1995), инициацию атеросклероза вызывают не столько липопротеины, сколько аутоиммунные комплексы, содержащие липопротеины в качестве антигена. Аутоиммунные комплексы имеют особенности:
вызывают повреждение эндотелия ускоряют проникновение липопротеинов в сосудистую стенку;
продлевают циркуляцию липопротеинов в крови и задерживают окисление и экскрецию холестерина с желчью способствуют развитию гиперлипопротеинемии;
откладываясь и фиксируясь в стенке артерий, оказывают цито-токсическое действие.
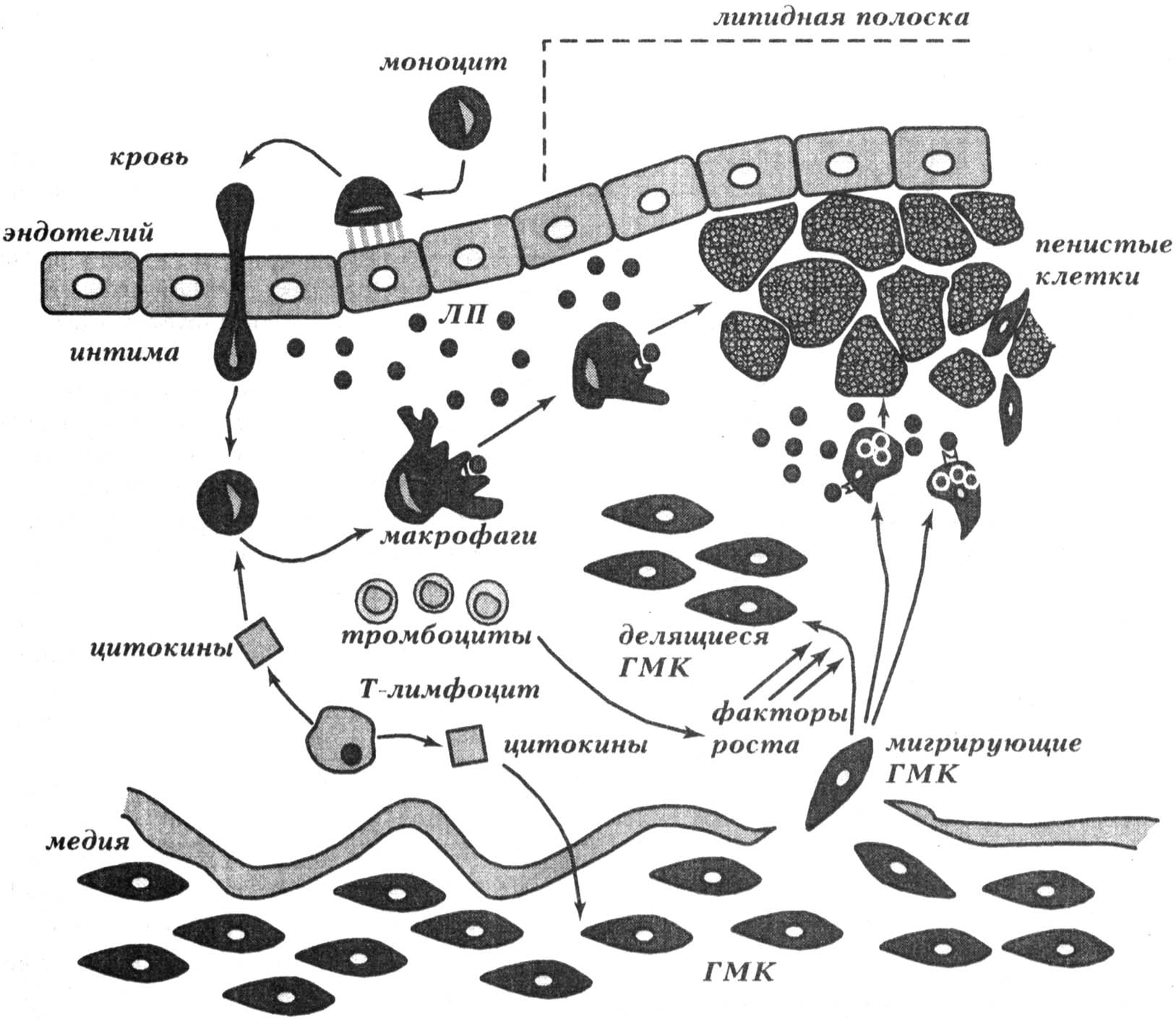
Рис. 15.16. Интегральные механизмы формирования ранних атеросклеротических поражений в артерии [по Д. Хайджару, А. Николсену, 1995]. ЛП – липопротеиды, ГМК – гладко-мышечные клетки.
Вирусная гипотеза - у больных атеросклерозом наблюдаются специфические изменения, характерные для инфекционного мононуклеоза, вызываемого вирусом Эпштейна-Барр. Причем только этот вирус обусловливает альтерацию эндотелия сосудов, пролиферацию гладкомышечных клеток и различные иммунопатологические сдвиги.
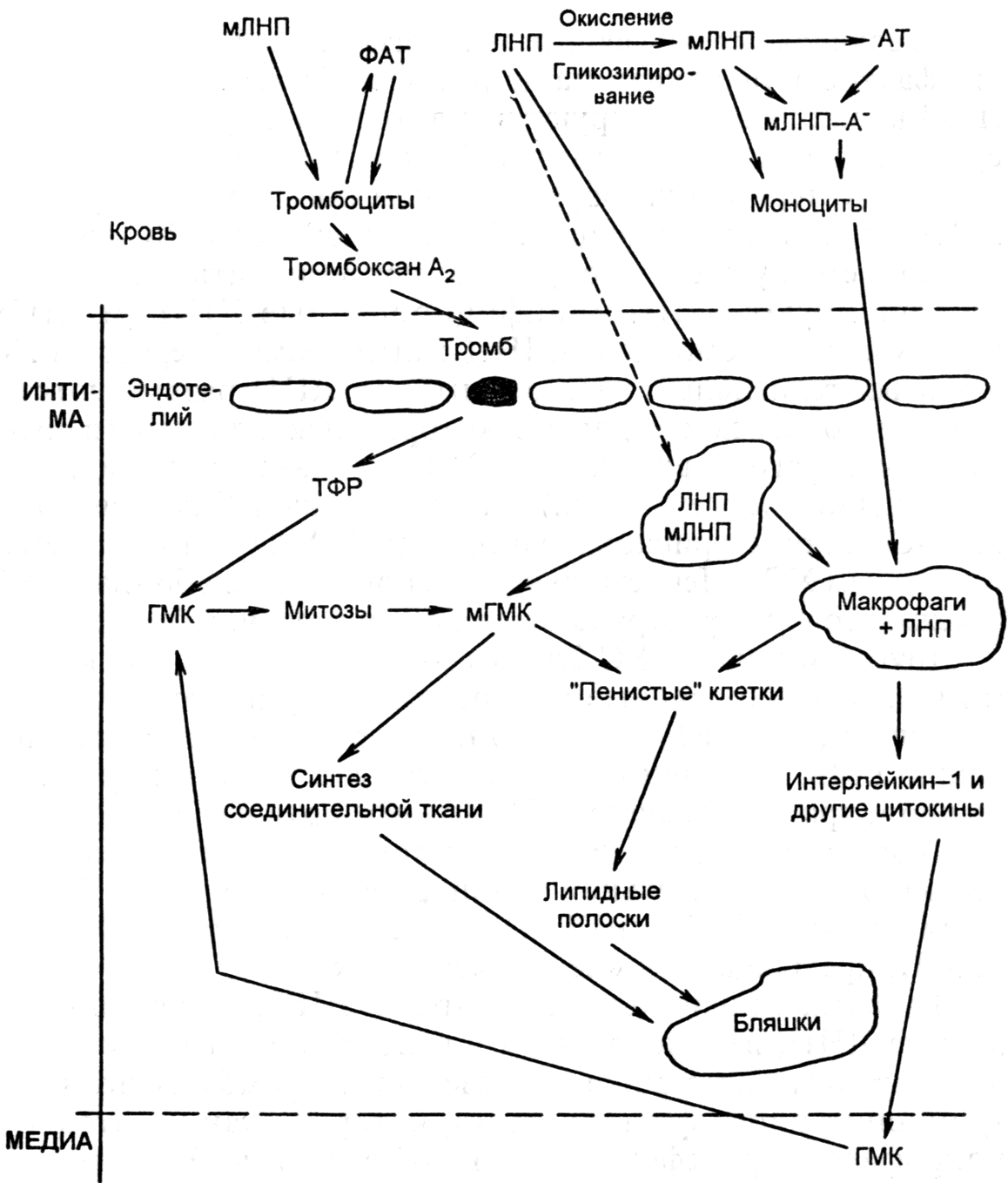
Рис. 15.17. Интегральная модель развития атеросклероза артериальной стенки [по Е.С. Северину, 2000]. ЛНП – липопротеины низкой плотности, мЛНП – модифицированные липопротеины низкой плотности, AT - антитела к мЛНП; ФАТ - фактор активации тромбоцитов; ТФР - тромбоцитариый фактор роста, ГМК – гладкомышечные клетки.
45. – Исходы и осложнения атеросклероза. Патогенез расстройств региональной гемодинамики при атеросклерозе.
46. – Патология белкового обмена. Нарушение усвоения белкаов пищи, белкового сотава плазмы крови и конечных этапов белкового обмена. Нарушение обмена аминокислот.
Нарушения поступления, переваривания и всасывания белков
Количественная или качественная белковая недостаточность первичного (экзогенного) происхождения, обусловленная ограничением поступления экзогенных белков при полном или частичном голодании, низкой биологической ценностью пищевых белков, дефицитом незаменимых аминокислот (валин, изолейцин, лейцин, лизин, метионин, треонин, триптофан, фенилаланин, гистидин, аргинин).
Квашиоркор - это тяжелое заболевание, вызванное белковым голоданием, квашиоркору нередко предшествует инфекция. Слово "квашиоркор" происходит из языка западно-африканского народа га (Ga), проживающего на территории современной Ганы. Оно означает "болезнь первенца после рождения младшего". После отнятия от груди первенец лишается источника полноценного белка и его питание, субдостаточное в калорическом отношении, становится, преимущественно, углеводистым. Это приводит к развитию характерной отёчной формы несбалансированной белково-энергетической недостаточности, сопровождаемой ранней потерей протеинов из висцерального пула организма.Основные симптомы - общая слабость, отеки, депигментация кожи. Замедление роста, анемия, гипопротеинемия, отек, жировая инфильтрация печени (дефицит ЛПОНП), атрофия поджелудочной железы, диарея, стеаторея. Уменьшение -глобулинов. Депигментация – следствие дефицита тирозина.
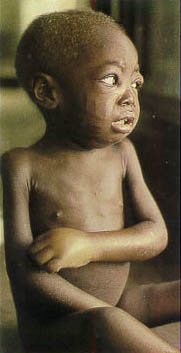
Рис.16.7. Ребенок с заболеванием квашиоркор, заметен периорбитальный отек, отек кистей рук и подкожной клетчатки живота, асцит, признаки депигментации (более светлая кожа) [http://med.fegi.ru/ris144.htm].
Алиментарный маразм (мумифицированная или сухая форма алиментарной дистрофии) - сбалансированная форма белково-энергетического дефицита. Характеризуется длительным компенсированным течением, когда нутриенты мобилизуются из соматического отсека тела, а висцеральный пул белка дольше сохраняется. При этом значительной степени достигает атрофия мышц и жировой клетчатки, но долго не возникает заметных отёков.
Дефицит пепсина может возникать при частичной резекции желудка за счет уменьшения секреции пепсиногена главными клетками слизистой (их количество сокращено), кроме того, при пониженной кислотности (низкое содержание соляной кислоты) пепсиноген плохо активируется до пепсина (при атрофическом гастрите). В результате этого белки не полностью расщепляются до пептидов, и вся нагрузка по их дальнейшему перевариванию ложится на тонкий отдел кишечника.
Дефицит трипсина, энтеропептидазы, карбоксипептидазы может быть, как результат смещения рН в более кислую сторону, патологии панкреаса (панкреатит, сдавление или закупорка протока) или нарушение секреции желудком гормона гастрина, который контролирует секрецию ферментов панкреаса. В результате негидролизованные белки и пептиды не могут всосаться в стенки кишечника и поступают в тонкий отдел, где подвергаются массовому гниению. При этом происходит аутоинтоксикация организма на фоне низкого содержания в крови аминокислот.
Нарушение работы гамма-глутамильного цикла всасывания аминокислот из-за дефицита любого фермента, катализирующего эти реакции.
Усиление перистальтики кишечника (энтероколиты) или уменьшение площади всасывания (оперативное удаление значительных участков тонкого кишечника) вызывает недостаточное действие пищеварительных ферментов на белки. Это ведет к резкому сокращению времени контакта содержимого химуса с апикальной поверхностью энтероцитов, незавершенности процессов энзиматического распада и процессов активного и пассивного всасывания.
Повреждение стенки тонкого кишечника (отек слизистой оболочки, воспаление) или неравномерное по времени всасывание отдельных аминокислот являются причинами нарушения всасывания аминокислот. Это ведет к нарушению (дисбалансу) соотношения аминокислот в крови и нарушению синтеза белка, поскольку незаменимые аминокислоты должны поступать в организм в определенных количествах и соотношениях. Чаще всего отмечается нехватка метионина, триптофана, лизина и ряда других аминокислот.
Нормализация переваривания и всасывания белков
Дефицит протеолитических ферментов желудка издавна компенсировался заместительной терапией желудочным соком животных. В случае большего поражения обкладочных клеток, для активации пепсиногена достаточно применять также в качестве заместительной терапии слабые растворы соляной кислоты, иногда применяют комплексные препараты (ацидин-пепсин, абомин, пепсидил). В случае гипосекреции желудка функционального, а не органического (атрофического) генеза, для рефлекторной стимуляции желудочной секреции применяют уже упоминавшиеся средства, раздражающие чувствительные окончания ротовой полости и желудка (например, горечи). Стимуляция секреции протеаз железами желудочно-кишечного тракта, а также поджелудочной железы возможна с помощью стимуляции активности центра блуждающего нерва или имитации его активности прямой (ацеклидин) или косвенной, блокируя холинэстеразу (прозерин, физостигмин) стимуляцией М-холинорецепторов желез.
Для заместительной фармакотерапии при дефиците протеаз кишечника в настоящее время применяют многочисленные комплексные ферментные препараты, биогенные препараты слизистой кишечника, поджелудочной железы (фестал, мезим, панзинорм, панкреатин).
В случае, когда усвоение (переваривание и всасывание) белков нарушено вследствие только усиленной перистальтики, кроме, естественно, борьбы с причиной (например, с помощью противомикробных препаратов - антибиотиков и сульфаниламидных препаратов "кишечной" группы) при инфекционных энтеритах, сорбентов при токсических энтеритах (энтеросорбент, карболонг, полифепан), для ослабления перистальтики применяют препараты, снижающие тонус или действие блуждающего нерва (средства, угнетающие центр вагуса, М-холиноблокаторы).
Нарушения белкового состава плазмы крови. Этиология.
Гипопротеинемия возникает, главным образом, за счет снижения количества альбуминов, синтезируемых печенью, а так же при протеинурии (потеря белков с мочой). Может быть приобретенной (при голодании, заболевании печени, почек, нарушении всасывания белков) и наследственной.
Гиперпротеинемия связана в основном с изменением содержания глобулинов за счет повышения гамма-глобулинов, синтезируемых плазматическими клетками (клетками иммунной системы), а также альфа- и бета-глобулинов, синтезирующихся печенью. Может быть относительной, при «сгущении» крови.
Парапротеинемия - появление измененных глобулинов. Например, при миеломной болезни они проходят почечный барьер и в моче определяются как белки Бенс-Джонсона.
Диспротеинемия - это нарушение соотношения альбуминов и глобулинов крови (альбумин-глобулиновый клэффициент).
Ликвидация гипопротеинэмии, в целом, проводится по тем же направлениям, что и коррекция отрицательного азотистого баланса. Учитывая, что основные клетки, которые синтезируют плазменные белки ‑ это гепатоциты и плазмоциты ("обученные" В-лимфоциты), требуется также коррекция их функции в случае нарушения (рассматривается в разделах "Патология печени" и "Иммунопатология"). Дефициты отдельных плазменных белков обычно не поддаются фармакологической коррекции. Заместительная терапия обычно проводится путем введения донорской плазмы или ее компонентов (см. терапию ДВС-синдрома, гемофилии, иммунодефицитов). Правда, как уже говорилось ранее, для усиления синтеза некоторых прокоагулянтов применяют введение кофермента, участвующего в этом процессе ‑ витамина К (викасол). В случае дефицита церулоплазмина при болезни Вильсона – Коновалова в качестве патогенетической терапии применяют введение соединений, связывающих ионы меди (пеницилламин). При необходимости активировать фибринолиз используют активаторы синтеза плазмина, при избытке ферментной активности плазмы крови (панкреатиты, ДВС-синдром), как указано в соответствующих разделах, используют антиферментные препараты.
Нарушение синтеза белка.
Нарушения синтеза белка приобретенного характера обусловлены преимущественно следующими причинами.
Различные виды алиментарной недостаточности (полное, неполное голодание, отсутствие в пище незаменимых аминокислот, нарушение определенного количественного соотношения между незаменимыми аминокислотами, поступающими в организм). Отсутствие в клетках хотя бы одной незаменимой аминокислоты прекращает синтез белка в целом.
Дефицит энергии при гипоксии.
Дефицит анаболических гормонов.
Избыток катаболических гормонов и факторов (ФНО при лихорадке и опухолях).
Опухоли.
Дефицит витаминов.
Нарушение иннервации вызывает дефицит трофогенов и развитие трофических язв.
Нарушение синтеза белка вызывают некоторые антибиотики. Так, "ошибки" в считывании генетического кода могут возникнуть под влиянием стрептомицина, неомицина и других антибиотиков. Тетрациклины тормозят присоединение новых аминокислот к растущей полипептидной цепи (образование прочных ковалентных связей между ее цепями), препятствуя расщеплению нитей ДНК.
Изменение активности ферментных систем клеток, участвующих в синтезе белка, носят в основном наследственный характер.
Гемоглобинопатии – аномалии, связанные с нарушением механизма синтеза белкового компонента гемоглобина при нормальной структуре гема. Выявлено более 15 видов аномальных молекул гемоглобина, где в альфа- или бета-цепи произошла замена одной из АК. Известно несколько видов мутантных гемоглобинов, где произошла замена остатка гистидина, который связывает железо гема с белковой частью молекулы, на другие АК. Получаются так называемые М-гемоглобины, которые не способны транспортировать кислород.
Глютеновая энтеропатия (целиакия) характеризуется нарушением всасывания вследствие иммуноопосредованного воспалительного повреждения слизистой оболочки тонкой кишки, пусковым фактором которого является потребление в пищу пшеничного глютена или родственных белков ржи и ячменя у генетически предрасположенных людей. Повреждающее действие на слизистую оболочку тонкой кишки оказывает глиадин - один из основных компонентов растительного белка глютена. Глиадин связывается со специфическим рецептором энтероцитов, взаимодействует с Т-лимфоцитами. Образующиеся лимфокины и антитела повреждают энтероциты ворсинок. На повреждающее действие глиадина слизистая оболочка отвечает атрофией и инфильтрацией иммунокомпетентными клетками.
Ранее считалось, что целиакия – болезнь генетической природы, при которой имеется дефицит ферментов, расщепляющих один из компонентов белка клейковины злаков до аминокислот, из-за чего в организме накапливаются продукты его неполного гидролиза.
Фенилкетонурия.
Альбинизм.
Алкаптонурия.
Наследственные энзимопатии, связанные с обменом аминокислот. Фармакологическая коррекция разных типов фенилкетонурии.
Таблица 16.1.
Наследственные энзимопатии, связанные с обменом аминокислот
Аминокислота |
Превращения в норме |
Ферментный дефицит |
Клинические проявления |
Прочее |
Фенилаланин |
Тирозин |
фенилаланингидроксилаза |
|
Диагностика: 5% трихлоруксусное железо → оливково-зеленое окрашивание мочи |
Тирозин |
Меланин Тироксин |
Тирозиназа, фермент йодирования |
|
|
Триптофан |
Серотонин |
|
|
|
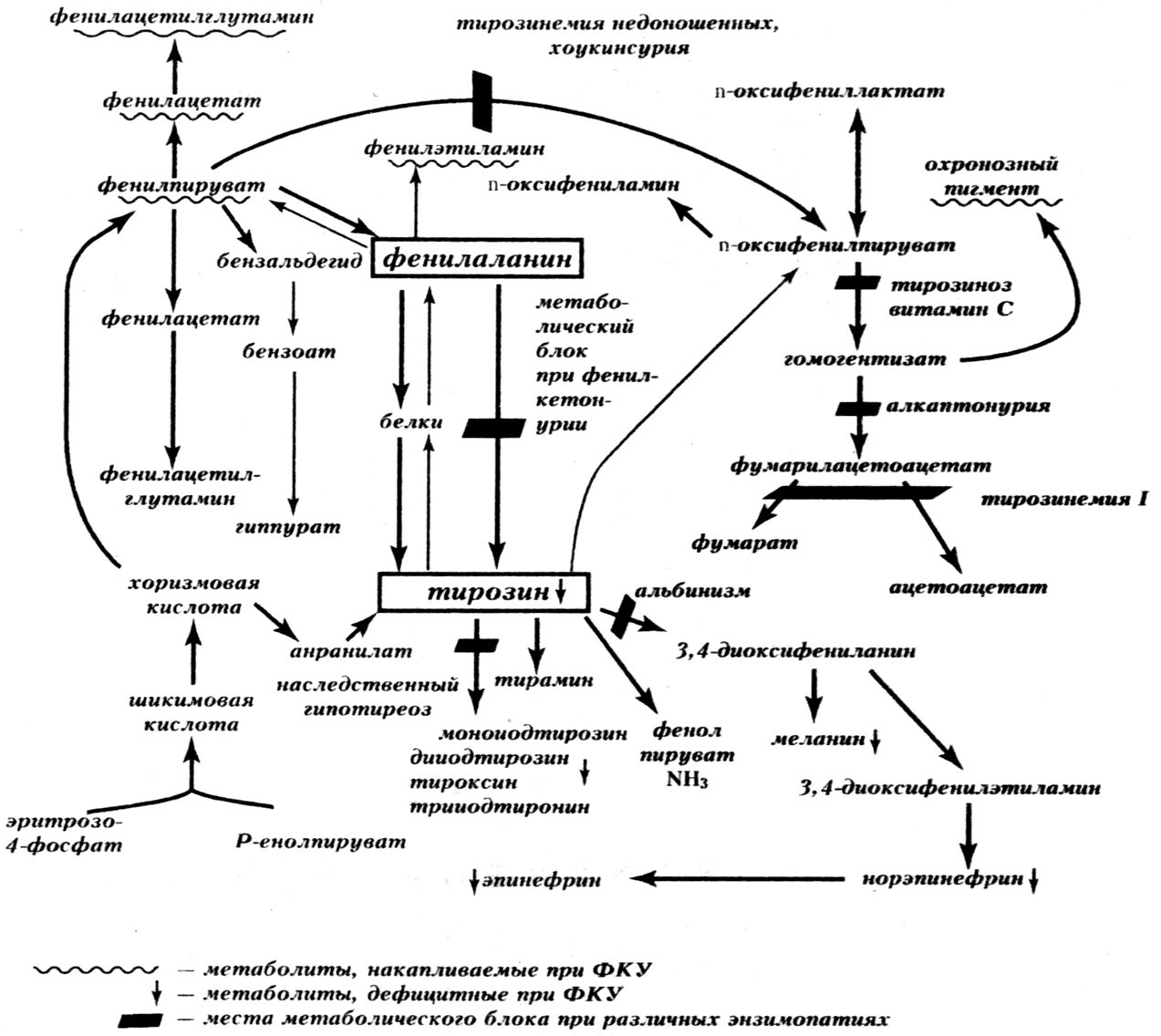
Рис. 16.8. Патогенез основных видов наследственных энзимопатий, связанных с обменом аминокислот [по А.Ш. Зайчику, Л.П. Чурилову, 2000].
Алкаптонурия – аутосомно-рецессивная болезнь, на примере которой сэр Арчибальд Гаррод (1909) создал концепцию метаболического блока. Причина заболевания - дефект оксидазы промежуточного продукта распада фенилаланина и тирозина - гомогентизиновой кислоты.
В норме фермент n-оксифенилпируватдезоксигеназа в присутствии витамина С, превращает полученный из тирозина n-оксифенилнируват в гомогентизиновую кислоту. Затем, гомогентизиновая кислота должна в почках окислиться в присутствии Fe+2 и глутатиона до 4-малеилацетоуксусной кислоты. Если этот процесс тормозится, то накопление гомогентизиновой кислоты ведет к ее превращению полифенолоксидазой в хиноновые полифенолы, составляющие так называемый «охронозный пигмент», выводимый почками. Это обусловливает потемнение мочи больных на воздухе.
Гомогентизиновая кислота ингибирует фермент лизилгидроксилазу, участвующий в синтезе коллагена, а охронозный пигмент (алкаптон) не полностью экскретируется с мочой, откладывается в основном веществе хрящей и других соединительно-тканных образований и делает их хрупкими, что со временем (после 30-ти лет) вызывает кальцификацию и дегенеративный артрит позвоночника, а также крупных суставов конечностей. Первыми проявлениями болезни могут быть пигментация склер и хрящей ушных раковин.
Радикально болезнь не лечится. Степень остеохондропатии можно уменьшить, защищая активность лизилгидроксилазы большими дозами аскорбиновой кислоты.
Фенилаланин - незаменимая аминокислота. Помимо включения в процесс биосинтеза белков, фенилаланин в печени метаболизируется двумя путями. В норме большая часть фенилаланина превращается в тирозин. Реакция катализируется фенилаланингидроксилазой при участии дигидробиоптерина.
Известны две гетерогенные группы наследственных нарушений обмена фенилаланина, сопровождающихся фенилаланинемией:
1) обусловленные дефектом фенилаланингидроксилазы,
2) являющиеся следствием дефектов ферментов, участвующих в синтезе и метаболизме тетрагидробиоптерина.
Фенилкетонурия (ФКУ) - наследственное заболевание, обусловленное мутациями в гене фенилаланингидроксилазы (классическая ФКУ).
Наиболее частая форма классической ФКУ проявляется характерным симптомокомплексом: умственная отсталость, судорожный синдром, склонность к экземе, нарушение пигментного обмена. От больных исходит специфический «мышиный» запах, обусловленный высоким содержанием фенольных кислот в биологических жидкостях. Эти симптомы появляются в первый год жизни, и больные при отсутствии лечения, как правило, не доживают до 30 лет.
В патогенезе симптомов ФКУ основную роль играют нарушения обмена циклических аминокислот:
• увеличение концентрации фенилаланина ограничивает транспорт тирозина и триптофана через гематоэнцефалический барьер (ГЭБ) и тормозит синтез нейромедиаторов;
• накопление фенилаланина в клетках мозга нарушает реакции синтеза цереброзидсульфатов, участвующих в защите мозга от демиелинизации;
• фенилаланин и его производные фенольные кислоты оказывают нейротоксическое действие. Они являются ингибиторами тирозингидроксилазы и триптофангидроксилазы - ключевых ферментов синтеза нейромедиаторов: дофамина, норадреналина, серотонина.
Для предотвращения токсического действия фенилаланина и фенилпирувата белковая пища просто заменяется набором аминокислот без фенилаланина.
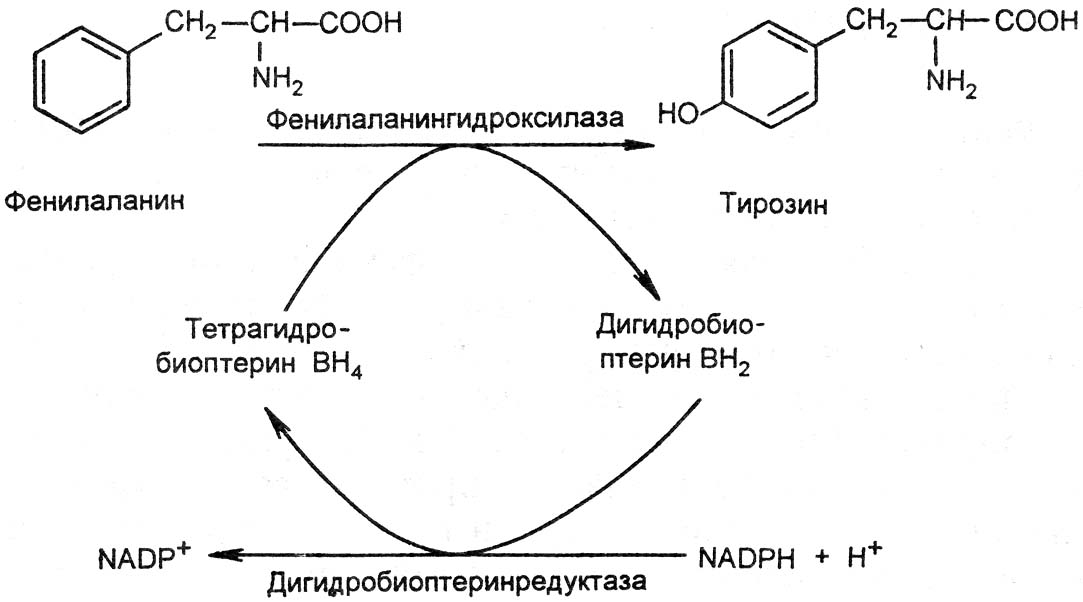
Рис. 16.9. Превращение фенилаланина в тирозин [по Е.С. Северину, 2000]. ВН4 синтезируется в организме из ГТФ, но в недостаточном количестве. Его регенерация при участии дигидробиоптеринредуктазы является необходимым звеном в превращении фенилаланина в тирозин.
Коферментзависимая гиперфенилаланинемия (вариантная ФКУ) является следствием мутаций в генах, контролирующих метаболизм тетрагидробиоптерина.
Тетрагидробиоптерин необходим в реакциях гидроксилирования не только фенилаланина, но также тирозина и триптофана. Поэтому при дефиците этого кофермента нарушается в равной степени метаболизм всех 3 аминокислот. Заболевание характеризуется тяжелой неврологической симптоматикой и ранней смертностью («злокачественная ФКУ»).
Данное заболевание поддается фармакотерапии тетрагидробиоптерином.
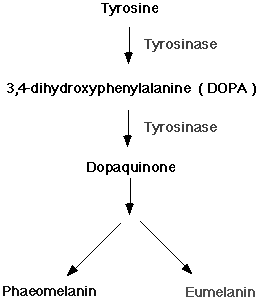
Рис. 16.10. Схема точек приложения тирозиназы [http://www.cnb.uam.es/~montoliu/ albinomouse.html]. Тирозиназа необходима для синтеза меланина в меланоцитах и пигментном эпителии сетчатки.
Альбинизм - врождённый дефицит или отсутствие пигмента в коже, волосах, радужке и сетчатке глаза или только в радужке глаза за счёт нарушения обмена тирозина при синтезе меланинов (отсутствие или дефицит тирозиназы). Принято различать генерализованные (кожно-глазные), изолированные (глазные) и смешанные типы альбинизма.
|
|

Рис. 16.11. Альбиносы на фотографиях IX века [http://members.optusnet.com.au/~msafier/albinism/]. Очаги альбинизма были выявлены в Северной Ирландии, в Южной Панаме. За цвет кожи и ночной образ жизни альбиносов называли «детьми Луны».
Нарушение обмена аминокислот. Патология конечных этапов белкового обмена, роль печени и почек в метаболизме аммиака. Нарушение обмена пуриновых и пиримидиновых оснований.
Нарушение переаминирования
Недостаточность в организме витамина В6 (фосфорилированная форма витамина В6 - фосфопиродоксаль - является активной группой трансаминаз - специфических ферментов переаминирования между амино- и кетокислотами). Беременность, голодание, заболевания печени, длительный прием сульфаниламидов оказывают тормозящий эффект на синтез витамина В6 и могут послужить основой нарушения обмена аминокислот.
Патологическое усиление реакции переаминирования возможно в условиях повреждения печени и инсулиновой недостаточности, когда значительно увеличивается содержание свободных аминокислот.
Нарушение синтеза трансаминаз (при белковом голодании).
Нарушение гормональной регуляции активности трансаминаз (глюкокортикоиды и тиреоидные гормоны стимулируют трансаминирование).
При некрозах отдельных органов (миокард, легкие, ПЖ, печень) наблюдается выход трансаминаз в кровь, что имеет диагностическое значение.
Нарушение дезаминирования
Ослабление может возникнуть вследствие нарушения окислительных процессов в тканях (гипоксия, гиповитаминозы С, В6 (пиродоксин), В2 (рибофлавин), РР (никотиновая кислота)).
Понижение активности аминооксидаз вследствие ослабления их синтеза (диффузное поражение печени, белковая недостаточность).
Понижение активности аминооксидаз вследствие относительной недостаточности их активности (увеличение содержания в крови свободных аминокислот).
Следствием нарушения окислительного дезаминирования аминокислот будет ослабление мочевинообразования, увеличение концентрации аминокислот (гипераминоацидемия) и увеличение выведения их с мочой (аминоацидурия).
Гипераминоацидемия - повышение концентрации аминокислот в крови.
Аминоацидурия - повышение содержания аминокислот в моче.
Патология конечных этапов белкового обмена
Азотистые продукты конечных этапов белкового обмена: мочевина, аммиак, мочевая кислота, креатинин, индикан.
Состав остаточного азота (20-30 мг%): на 50% состоит из азота мочевины, около 25% его приходится на долю аминокислот, остальная часть приходится на различные азотистые продукты.
Немочевинную часть называют резидуальным азотом.
Гиперазотемия - увеличение остаточного азота в крови.
Печеночная или продукционая азотемия связана с недостаточным образованием в печени мочевины. В этих случаях увеличивается количество резидуального азота.
Почечная или ретенционная азотемия обусловлена нарушением выделительной функции почек. Увеличивается содержание остаточного азота за счет азота мочевины.
Токсичность аммиака. Повышение концентрации аммиака в плазме крови снижает фонд метаболитов и АТФ в клетках ЦНС.
Один из важнейших метаболитов цитратного цикла -кетоглутарат в глутаматдегидрогеназной реакции превращается в глутаминовую кислоту. Глутамат в свою очередь используется для синтеза глутамина. Накопление глутамина в клетках нейроглии приводит к повышению в них осмотического давления и набуханию астроцитов. Кроме того тормозится цикл Кребса в ЦНС за счет расходования -кетоглутарата.
Аммиак в крови при рН=7,4 существует почти целиком в виде NH4+. Ионы аммония проникают через плазматическую и митохондриальную мембраны с большим трудом. Повышение концентрации NH4+ создает препятствие для трансмембранного переноса других ионов и приводит к сдвигу рН. NH3 составляет при рН 7,4~1 %, но он легко проходит через мембраны клеток и в митохондриях сдвигает равновесие глутаматдегидрогеназной реакции в сторону образования глутамина. Изменение метаболизма глутамата в сторону образования глутамина нарушает процесс синтеза важнейшего тормозного медиатора ЦНС -аминомасляной кислоты.
Нарушение метаболизма аминокислот в печени приводит к повышению концентрации в крови циклических аминокислот и жирных кислот с короткой цепью, что в свою очередь оказывает токсическое воздействие на клетки мозга.
47. – Дисгидрии. Основные виды. Этиология и патогенез. Несахарные диабеты.
Единой общепринятой классифиации нарушений водно-электролитного баланса не существует. Прежде всего, принято делить эти нарушения в зависимости от изменений объема воды:
Положительный водный баланс:
гипергидратация;
отеки.
Отрицательный водный баланс (гипогидратация).
Каждая из форм подразделяется на:
Экстрацеллюлярную (например, при гипоальдостеронизме гипоосмолярная гипогидратация).
Интрацеллюлярную;
Тотальную (например, при сахарном диабете гиперосмолярная гипогидратация).
Комбинированную, когда в одном секторе гипергидратация, в другом гипогидратация (например, при употреблении алкоголя тормозится выработка АДГ (отсюда - полиурия), и развивается внутриклеточная гипогидратация (жажда, соль) и внеклеточная – гипергидратация (с отеками).
В зависимости от осмотической концентрации гипер- и гипогидратации подразделяют на:
изоосмолярную;
гипоосмолярную;
гиперосмолярную.
Гипогидратация (дегидратация или эксикоз) - недостаточное поступление либо избыточное выделение жидкости.
Изоосмолярная возникает при острой кровопотере, плазмопотерях при обширных ожогах, т.е. когда теряется изотоническая жидкость.
Гиперосмолярная развивается в результате потери воды, превышающей ее поступление и эндогенное образование (потеря воды происходит с небольшой потерей электролитов) при сильном потении, гипервентиляции, поносе, полиурии, если утраченная жидкость не компенсируется питьем. Большая потеря воды с мочой бывает при так называемом осмотическом диурезе.
Гиперосмолярная дегидратация значительно легче возникает у грудных детей, чем у взрослых. В грудном возрасте большие количества воды почти без электролитов могут теряться через легкие при лихорадке, умеренном ацидозе и в других случаях гипервентиляции. Кроме того, у грудных детей недостаточно развита концентрационная способность почек. Преобладание потери воды над выделением электролитов приводит к увеличению осмотической концентрации внеклеточной жидкости и передвижению воды из клеток в экстрацеллюлярное пространство, что приводит к обезвоживанию клеток, которое вызывает мучительное чувство жажды, усиление распада белка, повышение температуры.
Гипоосмолярная развивается в тех случаях, когда организм теряет много жидкости, содержащей электролиты, а возмещение потери происходит меньшим объемом воды без введения соли, при повторной рвоте, поносе, сильном потении, полиурии (несахарный и сахарный диабет, патология почек), если потеря воды (или гипотонических растворов) частично восполняется питьем без соли.
Из гипоосмотического внеклеточного пространства часть жидкости устремляется в клетки, что приводит к развитию внутриклеточного отека. Чувство жажды при этом отсутствует. Потеря воды сопровождается нарастанием гематокрита, что приводит к повышению вязкости крови и нарушения микроциркуляции. Уменьшение объема циркулирующей крови ведет к уменьшению минутного объема сердца, а, следовательно, и к экстраренальной почечной недостаточности. Объем фильтрации резко падает, развивается олигурия.
Гипергидратация - избыточное поступление либо недостаточное выделение жидкости.
Изосмолярная возникает при вливании больших количеств физиологического или рингеровского раствора в эксперименте или больным в послеоперационном периоде.
Изоосмолярная гипергидратация не вызывает перераспределения жидкости между внутри- и внеклеточными фазами, осмотические свойства которых не изменены. Увеличение общего объема воды в теле совершается за счет внеклеточной жидкости, отсюда развитие гипертензии.
Гипоосмолярная (или водное отравление) возникает при избыточном накоплении воды без соответствующей задержки электролитов.
при проведении перитонеального диализа против гипоосмотического раствора, когда поступление воды превосходит способность почек к ее выделению, что имеет место при повышенной продукции АДГ (синдром Пархона, разбавленной гипонатрийемии) или олигоанурии (при острой почечной недостаточности).
в результате внутривенного вливания больших количеств изотонического раствора глюкозы, которая быстро потребляется клетками.
При водном отравлении вначале падает осмотическая концентрация внеклеточной жидкости благодаря ее разведению избытком воды. Осмотический градиент между "интерстицием" и клетками обуславливает передвижение части межклеточной воды в клетки и их набухание. Объем клеточной воды может повышаться на 15%.
Гиперосмолярная возникает при введении гипертонических растворов в объемах, превышающих возможность достаточно быстрого выделения их почками, например, при вынужденном питье морской воды. При этом происходит передвижение воды из клеток во внеклеточное пространство, ощущаемое как тяжелое чувство жажды.
Дифференциальная диагностика различных типов дисгидрий представлена в табл. 17.1.
Таблица 17.1.
Дифференциальная диагностика типов дисгидрий.
Тип дисгидрии |
ОЦК |
Осмотическое давление плазмы |
Содержание Na+ в плазме |
Дегидратация гипотоническая |
↓ |
↓ |
↓ |
Дегидратация изотоническая |
↓ |
N |
N |
Дегидрадация гипертоническая |
↓ |
↑ |
↑ |
Гипергидратация гипотоническая |
↑ |
↓ |
↓ |
Гипергидратация изотоническая |
↑ |
N |
N |
Гипергидратация гипертоническая |
↑ |
↑ |
↑ |
Примечание. ↑- повышено; ↓- снижено; N - в норме
48. – Этиология и патогенез отеков.
Отек – типовой патологический процесс, характеризующийся избыточным накоплением жидкости в межклеточном пространстве, в результате нарушения обмена между плазмой крови и периваскулярной жидкостью.
Анасарка – отек подкожной клетчатки (рис. 17.5.).
Рис. 17.5. Вид передней брюшной стенки при анасарке [http://www.lf2.cuni.cz/Projekty/interna/foto/010/pic00053.jpg].
Водянка – скопление внеклеточной жидкости в серозных полостях (асцит - водянка брюшной полости (рис. 17.6.), гидроторакс - водянка плевральной полости, водянка яичка - водянка скопление жидкости между оболочками яичка).
Рис. 17.6. Вид больной с асцитом при циррозе печени. [http://www.murrasaca.com/Hepaticirrosis.htm].
Транссудат – отечная жидкость невоспалительного характера.
Экссудат – отечная жидкость воспалительного характера.
Общие механизмы развития отеков:
Повышение гидростатического давления в венозном отделе капилляра.
Понижение коллоидно-осмотического давления плазмы крови, и прежде всего развитие гипопротеинемии.
Снижение механического противодавления ткани процессу фильтрации, наступающее при ее разрыхлении.
Повышение онкотического и осмотического давления интерстициальной жидкости, а также усиление способности белков к связыванию воды (набуханию).
Повышение проницаемости гемато-паренхиматозного барьера.
Нарушение оттока лимфы.
Нарушение нейро-эндокринной регуляции функции почек, и, прежде всего нарушение регуляции экскреции натрия почками.
Общий механизм отеков в соответствии с законом Франка-Старлинга (рис. 17.7.)
ФД = (ГДК + ОДТ) - (ГДТ + ОДК) |
КРОВЬ
35
25
капилляры
венулы
ТКАНЬ
15
ФДа=(32,5+4,5)-(3+25)=9
mmHg
ФДв=(17,5+4,5)-(3+25)=-6
mmHg
артериолы
17,5





Рис. 17.7. Общий механизм отеков в соответствии с законом Франка-Старлинга. ГДК - гидростатическое давление крови (32,5-17,5 мм рт.ст. на артериальном и венозном конце капилляров, соответственно). ОДТ - онкотическое давление ткани (4,5 мм рт.ст). ГДТ - гидростатическое давление ткани (3 мм рт.ст). ОДК - онкотическое давление крови (25 мм рт.ст.).
Патогенетическая классификация отеков
Гемодинамический отек возникает вследствие повышения давления крови в венозном отделе капилляров, что уменьшает величину реабсорбции жидкости при продолжающейся ее фильтрации (сердечная недостаточность, недостаточность клапанов вен (рис. 17.8.), венозный тромбоз).
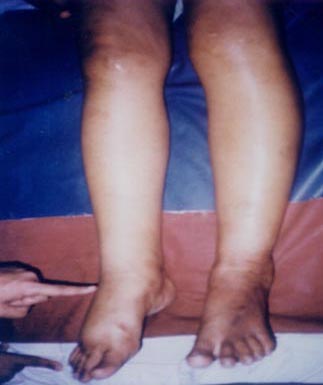
Рис. 17.8. Нижние конечности больной с гемодинамическими отеками [http://www.bhj.org/journal/2002_4401_jan/case_113.htm].
Онкотический отек развивается вследствие либо понижения онкотического давления крови, либо повышения его в межклеточной жидкости. Гипоонкия крови чаще всего бывает обусловлена снижением уровня белка и главным образом альбуминов. Гипопротеинемия может возникнуть в результате: недостаточного поступления белка в организм; нарушения синтеза альбуминов при заболеваниях печени; черезмерной потери белков плазмы крови с мочой при некоторых заболеваниях почек. Гиперонкия тканей может возникнуть в результате альтерации, нарушения проницаемости клеточных мембран.
Мембраногенный отек формируется вследствие значительного возрастания проницаемости сосудистой стенки при воспалении, действии токсинов, аллергических реакциях.
Осмотический отек может возникать и вследствие понижения осмотического давления крови или повышения его в межклеточной жидкости. Принципиально гипоосмия крови может возникать, но быстро формирующиеся при этом тяжелые расстройства гомеостаза "не оставляют" времени для развития его выраженной формы. Гиперосмия тканей, как и гиперонкия их, чаще носит ограниченный характер. Она может возникать вследствие: нарушения вымывания электролитов и метаболитов из тканей при нарушении микроциркуляции; снижения активного транспорта ионов через клеточные мемраны при тканевой гипоксии; массивной "утечки" ионов из клеток при их альтерации; увеличения степени диссоциации солей при ацидозе.
Лимфогенный отек формируется при повышении давления в лимфатических сосудов и возрастании проницаемости их стенки (рис. 17.9.). Длительно существующий лимфогенный отек сопровождается разрастанием соединительной ткани и развитием слоновости.
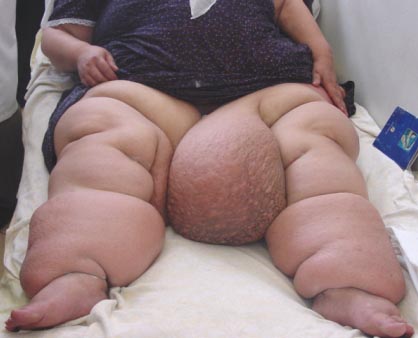
Рис. 17.9. Выраженный лимфогенный отек нижних конечностей [http://www.surgical-tutor.org.uk/default-home.htm?system/vascular/lymphoedema.htm~right]
Этиологическая классификация отеков
(в скобках указан патогенетический механизм отека)
Сердечный (центральный гемодинамический, застойный), см. рис. 17.10.
Венозный (периферический гемодинамический).
Почечный (нефритический, нефротический).
Эндокринный (при альдостеронизме).
Голодный (онкотический).
Кахектический (онкотический).
Печеночный (онкотический).
Воспалительный (мембраногенный).
Аллергический (мембраногенный).
Токсический (мембраногенный).
Лимфатический (с развитием слоновости).
Нейрогенный (питьевой).
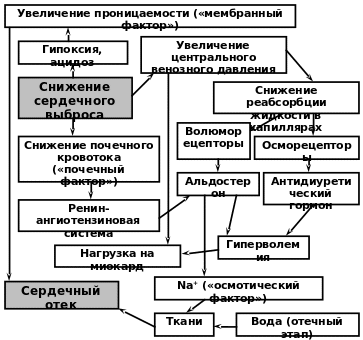

Рис. 17.10. Схема патогенеза сердечных отеков.
Нефритический отек (поражение клубочков)
Основную роль при поражении клубочкового аппарата почки играет активация ренин-ангиотензин-альдостероновой системы, следствием которой является задержка натрия и воды. Кроме этого определенную роль играет также повышение проницаемости клубочков из-за их иммунокомплексного повреждения (при гломерулонефрите, системной красной волчанке).
Важная роль принадлежит гиалуронидазе. Гиалуронидаза («фактор проницаемости») является ферментом микробного или тканевого происхождения, обладающим специфическим свойством - вызывать деполимеризацию и гидролиз гиалуроновой кислоты (входящей в состав межклеточного вещества стенки сосудов и капилляров). При некоторых заболеваниях ее активность в сыворотке крови возрастает (у больных с острым и обострением хронического гломерулонефрита, с нефротическим синдромом). Это вызывает деполимеризацию гиалуроновой кислоты, и сопровождается увеличением размеров микропор в сосудистой и капиллярной стенках, через которые усиливается уход в ткани не только воды и ионов (в том числе и ионов натрия), но и мелкодисперсных фракций белка (альбуминов). Таким образом, основное вещество соединительной ткани приобретает резко выраженные гидрофильные свойства, что также играет существенную роль в развитии отеков.
Нефротический отек (поражение турбулярного аппарата)
Поражение канальцевого аппарата почки сопровождается гиперпротеинурией, а ее следствием является гипоонкия плазмы. Снижение онкотического давления крови неизбежно приводит к потере ее жидкой части, т.е. развивается гиповолемия раздражение осмо- и волюморецепторов гипоталамуса. Возбуждение осморецепторов – стимул для выброса АДГ, волюморецепторов – альдостерона. Оба гормона способствую задержке воды.
49. – Нарушение солевого обмена. Основные причины и проявления нарушения обмена Na, K, Ca и Р.
Минеральный обмен – совокупность процессов всасывания, распределения, усвоения и выделения минеральных веществ, находящихся в организма преимущественно в виде неорганических соединений.
По количественному содержанию в организме они делятся на макроэлементы, если их больше чем 0,01 % от массы тела (К, Са, Мg, Na, P, Cl) и микроэлементы (Mn, Zn, Cr, Cu, Fe, Co, Al, Se). Основную часть минеральных веществ организма составляют хлористые, фосфорнокислые и углекислые соли натрия, кальция, калия, магния. Соли в жидкостях организма находятся в частично или полностью диссоциированном виде, поэтому минеральные вещества присутствуют в виде ионов – катионов и анионов.
Функции минеральных веществ:
пластическая (кальций, фосфор, магний);
поддержание осмотического давления (калий, натрий, хлор);
поддержание буферности биологических жидкостей (фосфор, калий, натрий);
поддержание коллоидных свойств тканей (все элементы);
детоксикационная (железо в составе цитохрома Р-450, сера в составе глутатиона);
проведение нервного импульса (натрий, калий);
участие в ферментативном катализе в качестве кофактора или ингибитора;
участие в гормональной регуляции (йод, цинк и кобальт входят в состав гормонов).
Промежуточный и конечный обмен минеральных веществ
Поступают минеральные вещества в организм в свободном или связанном виде. Ионы всасываются уже в желудке, основная часть минеральных веществ – в кишечнике путем активного транспорта при участии белков – переносчиков. Из желудочно-кишечного тракта поступают в кровь и лимфу, где связываются со специфическими транспортными белками. Выделяются минеральные вещества главным образом в виде солей и ионов.
С мочой: натрий, калий, кальций, магний, хлор, кобальт, йод, бром, фтор.
С калом: железо, кальций, медь, цинк, марганец, молибден, и тяжелые металлы.
Характеристика отдельных элементов
Натрий – основной (“большой”) катион внеклеточной жидкости. Составляет 0,08 % от массы тела, 135-150 мМ/л плазмы. Играет главную роль в поддержании осмотического давления. При отсутствии или ограничении в поступлении натрия в организм его выделение с мочой почти полностью прекращается. Всасывается в верхнем отделе тонкого кишечника при участии белков-переносчиков и требует затраты АТФ. Суточная потребность варьирует в зависимости от водно-солевого обеспечения организма. Депонируется в коже и мышцах. Кишечная потеря натрия происходит при диареях.
участвует в возникновении и поддержании электрохимического потенциала на плазматических мембранах клеток;
регулирует состояние водно-солевого обмена;
участвует в регуляции работы ферментов;
компонент K+-Na+ насоса.
Калий – составляет 0,25% от массы тела. Во внеклеточном пространстве содержится только 2% от общего количества (в плазме 3,5-5,5 мМ/л), а остальное - в клетках (150 мМ/л), где связан с углеводными соединениями. Всасывается на протяжении всего желудочно-кишечного тракта. Часть калия откладывается в печени и коже, а остальная поступает в общий кровоток. Обмен очень быстро протекает в мышцах, кишечнике, почках и печени. В эритроцитах и нервных клетках более медленный обмен калия. Играет ведущую роль в возникновении и проведении нервного импульса. Необходим для синтеза белков (на 1 г белка – 20 мг ионов калия), АТФ, гликогена, принимает участие в формировании потенциала покоя. Выделяется в основном с мочой и меньше с калом.
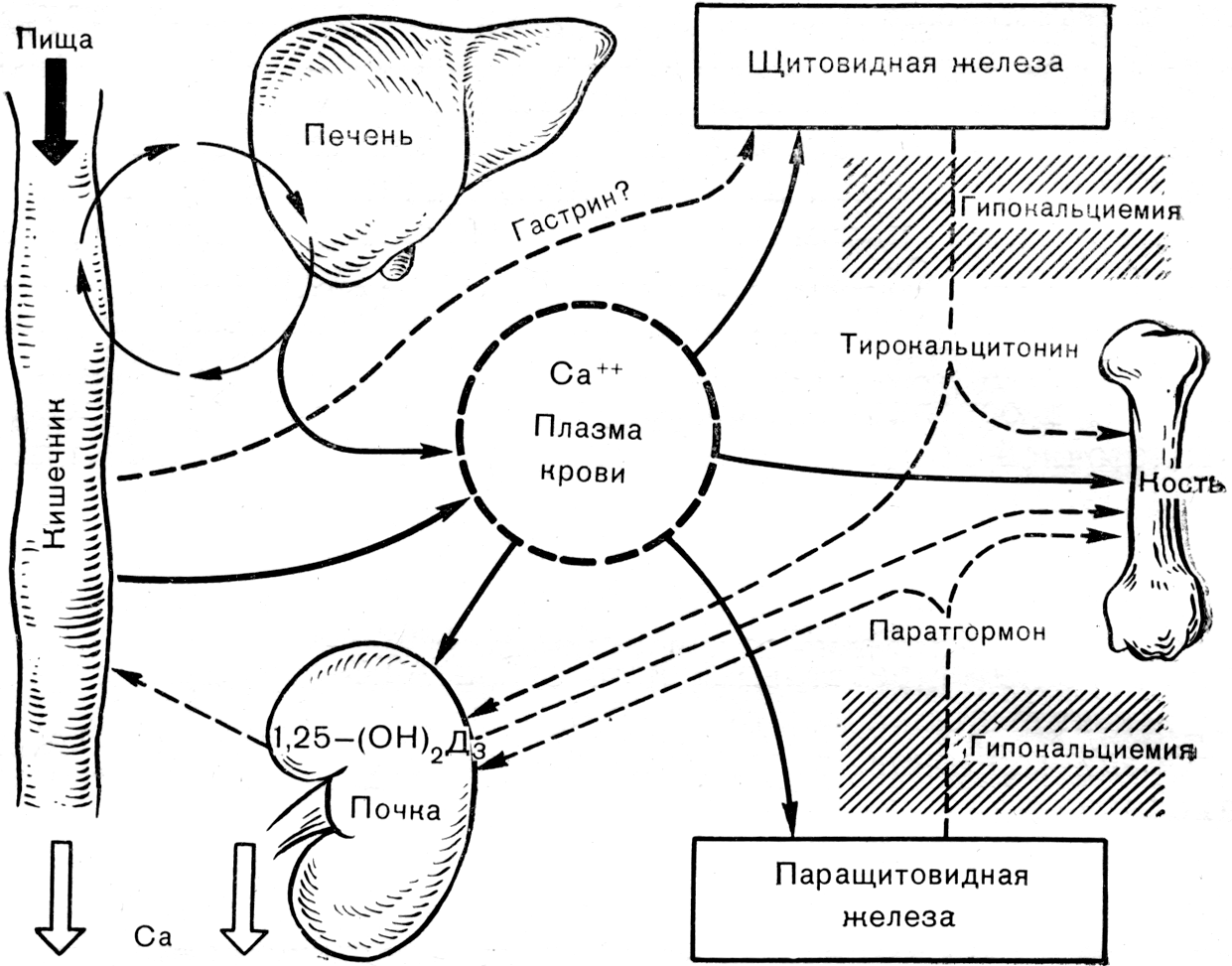
Рис. 17.11. Схема регуляции баланса кальция [по Тарееву Е.М., 1983].
Кальций – внеклеточный катион. Составляет 1,9 % от массы тела, 2,1-2,65 мМ/л плазмы. Содержание повышается в период роста или беременности. Функционирует как составная часть опорных тканей или мембран, участвует в проведении нервного импульса и инициации мышечного сокращения, является одним из факторов гемокоагуляции. Обеспечивает целостность мембран (влияет на проницаемость), т. к. способствует плотной упаковке мембранных белков. Кальций ограничено участвует в поддержании осмотического равновесия. Вместе с инсулином активирует проникновение глюкозы в клетки. Всасывается в верхнем отделе кишечника. Степень его усвоения зависит от рН среды (соли кальция в кислой среде нерастворимы). Жиры и фосфаты препятствуют всасыванию кальция. Для полного усвоения из кишечника необходимо наличие активной формы витамина D3. Схема регуляции баланса кальция показана на рис. 17.11.
Витамин D3 увеличивает синтез кальций-связывающего белка в энтероцитах, увеличивает содержание в энтероцитах кишки фосфолипидов, что повышает ее проницаемость для кальция, стимулирует рост и дифференцировку энтероцитов (что увеличивает всасывание, как кальция, так и фосфора).
Синтез витамина D3 происходит в коже под действием ультрафиолетового излучения. Сначала образуется провитамин D3, под действием тепла в коже происходит его изомеризация в витамин D3, поступает в печень, там происходит его гидроксилирование, затем поступает в почки, где происходит еще одно гидроксилирование и образуется активная форма. В почках этот процесс регулирует паратгормон, женские и мужские половые гормоны, гормон роста – стимуляторы образования витамина D3.
Большая часть кальция содержится в костной ткани (99%) в составе микрокристаллов карбонатапатита 3Са2(РО4)2 СаСО3 и гидроксилапатита 3Са2(РО4)2 СаОН.
Общий кальций крови включает три фракции: белоксвязанный, ионизированный и неионозированный (который находится в составе цитрата, фосфата и сульфата).
Неорганический фосфор - содержится преимущественно в костной ткани. Составляет 1% от массы тела. В плазме крови при физиологических рН фосфор на 80 % представлен двухвалентным и на 20 % одновалентным анионом фосфорной кислоты. Фосфор входит в состав коферментов, нуклеиновых кислот, фосфопротеинов, фосфолипидов. Вместе с кальцием фосфор образует апатиты – основу костной ткани.
Магний – составляет 0,05% от массы тела. В клетках его содержится в 10 раз больше, чем во внеклеточной жидкости. Многого магния в мышечной и костной ткани, также в нервной и печеночной. Образует комплексы с АТФ, цитратом, рядом белков.
входит в состав почти 300 ферментов;
комплексы магния с фосфолипидами снижают текучесть клеточных мембран;
участвует в поддержании нормальной температуры тела;
участвует в работе нервно-мышечного аппарата.
Хлор – важнейший анион внеклеточного пространства. Составляет 0,06% от массы тела. Большая часть его содержится в желудочном соке. Участвует в поддержании осмотического равновесия. Активирует амилазу и пептидазы. Всасывается в верхних отделах кишечника, выделяется в основном с мочой. Концентрация хлора и натрия обычно изменяются параллельно.
Медь входит в состав многих ферментов и биологически активных металлопротеинов. Участвует в синтезе коллагена и эластина. Является компонентом цитохрома с электронтранспортной цепи.
Сера – составляет 0,08%. Поступает в организм в связанном виде в составе АК и сульфат-ионов. Входит в состав желчных кислот и гормонов. В составе глутатиона участвует в биотрансформации ядов.
Железо входит в состав железосодержащих белков и гема гемоглобина, цитохромов, пероксидаз.
Цинк – является кофактором ряда ферментов.
Кобальт входит в состав витамина В12.
Таблица 17.2.
Нарушения минерального обмена
Тип |
Основные причины |
Проявления |
Коррекция |
Гипернатрийемия (более 150 мМ/л) |
|
Увеличение нервно-мышечной возбудимости, судороги, гипертензия, отеки. |
0,9%-ный р-р NaCl, 5%-ный р-р глюкозы, гемодиализ. |
Гипонатрийемия |
|
Снижение нервно-мышечной возбудимости, миастения, тахикардия, гипотония. |
1-2%-ный р-р NaCl, плазма, белковые препараты. |
Гиперкалийемия (более 5,5-8 мМ/л) |
|
Мышечные параличи, брадикардия, аритмия, расширение QRS и Т на ЭКГ, ацидоз, остановка сердца в фазу диастолы. |
Р-р глюкозы с инсулином.
|
Гипокалийемия (менее 3,5-6 мМ/л) |
|
Снижение нервно-мышечной возбудимости (из-за гиперполяризации), миастения, атония кишечника, аритмии, удлинение интервалов PQ, QT, уплощение и инверсия Т на ЭКГ (нарушения процессов реполяризации), снижение секреции соляной кислоты, алкалоз. |
Патогенетически, р-р KCl. |
Гиперкальцийемия, гипофосфатемия |
|
Жажда, полиурия, диспепсия, гипотония мышц, гиперрефлексия, гипертензия, остеопороз. |
Na2 ЭДТА |
Гипокальцийемия и гиперфосфатемия |
|
Повышение нервно-мышечной возбудимости, тетанические судороги, гипокоагуляция, гипотензия, рахит, катаракта. |
Заместительная гормонотерапия, CaCl2, глюконат и лактат кальция, витамин D. |
Гипермагнийемия |
|
Угнетение ЦНС, нарушение дыхания, брадикардия, гипотония. |
Бикарбонат и лактат натрия. |
Гипомагнийемия |
|
Повышение нервно-мышечной возбудимости, гиперрефлексия, тремор, тетания, тахикардия, гипертония. |
Соли магния, патогенетически. |
Изменение нервно-мышечной возбудимости при нарушении минерального обмена определяется формулой, в числителе которой - сумма ионов Na+, K+, бикарбоната и фосфата, а в знаменателе – Ca++, Mg++ и H+. Рост числителя и уменьшение знаменателя повышает нервно-мышечную возбудимость.
50. – Нарушения кислотно-основного состояния (КОС). Роль легких, почек, печени, ЖКТ в регуляции КОС в норме и патологии. Основные формы и проявления нарушений (КОС).
Кислотно-основное состояние (КОС) – соотношение между активными массами водорода и гидроксильных ионов.
Физиологические системы регуляции КОС
Легкие - это первая линия защиты в поддержании кислотно-основного гомеостаза, поскольку они обеспечивают механизм почти немедленной регуляции выделения кислоты. В то же время любые нарушения дыхания, сопровождающиеся увеличением или уменьшением минутной альвеолярной вентиляции, могут стать причиной развития нарушений КОС.
Вдыхаемый воздух содержит незначительное количество СО2. Почти вся углекислота крови является продуктом клеточного метаболизма. По мере образования в процессе клеточного метаболизма СО2 легко диффундирует в капилляры и транспортируется к легким в трех основных формах:
растворенный СО2;
анион бикарбоната;
карбаминовое соединение.
СО2 очень хорошо растворяется в плазме. Около 5 % общей двуокиси углерода в артериальной крови находится в форме растворенного газа, а 90 % - в форме бикарбоната. Последний является продуктом реакции СО2 с водой с образованием Н2СО3 и ее диссоциацией на водород и ион бикарбоната:
карбоангидраза
СО2 + Н2О Н2СО3 Н+ + НСО3-.
Реакция между СО2 и Н2О протекает медленно в плазме и очень быстро в эритроцитах, где присутствует внутриклеточный фермент карбоангидраза. Она облегчает реакцию между СО2 и Н2О с образованием Н2СО3.
По мере накопления НСО3- внутри эритроцита анион диффундирует через клеточную мембрану в плазму. Мембрана эритроцита плохо проницаема для Н+, как и вообще для катионов, поэтому ионы водорода остаются внутри клетки. Электрическая нейтральность клетки в процессе диффузии НСО3- в плазму обеспечивается потоком ионов хлора из плазмы в эритроцит.
Часть Н+, остающихся в эритроцитах, соединяется с гемоглобином. В периферических тканях, где концентрация СО2 высока и значительное количество Н+ накапливается эритроцитами, связывание Н+ облегчается деоксигенацией гемоглобина. Восстановленный гемоглобин лучше связывается с Н+, чем оксигенированный. Таким образом, деоксигенация артериальной крови в периферических тканях способствует связыванию Н+ посредством образования восстановленного гемоглобина. Это увеличение связывания СО2 с гемоглобином известно как эффект Холдейна.
Третьей формой транспорта СО2 являются карбаминовые соединения, образованные в реакции СО2 с концевыми аминогруппами белков крови. Основным белком крови, связывающим СО2 является гемоглобин. Этот процесс описывается реакцией:
Hb-NH2 + СО2 Hb-NH СООН Hb-NHCOO- + H+.
Реакция СО2 с аминогруппами протекает быстро. Как и в случае более легкого связывания СО2 с восстановленным гемоглобином, образование карбаминовых соединений легче протекает с деоксигенированной формой гемоглобина. Карбаминовые соединения составляют около 5 % общего количества СО2, транспортируемого артериальной кровью.
Регуляция выделения СО2 достигается изменением скорости объема легочной вентиляции. Повышение вентиляции приводит к снижению артериального рСО2 и наоборот. Афферентные сигналы, изменяющие альвеолярную вентиляцию, связаны с хеморецепторами, которые регулируют функции дыхательного центра. Эти рецепторы находятся в продолговатом мозге, аортальном и каротидном тельцах и реагируют на изменения рСО2 и концентрации Н+.
При увеличении кислотности крови, повышение содержания ионов H+ приводит к возрастанию легочной вентиляции (гипервентиляции). Н+ связываются с NaHCO3 с образованием H2CO3. H2CO3 разлагается на CO2 и H2O. При этом молекулы CO2 выводятся в большом количестве и pH возвращается к нормальному уровню.
При увеличении содержания оснований наступает гиповентиляция, в результате напряжение CO2 и концентрация ионов H+ возрастают, и сдвиг реакции крови в щелочную сторону частично или полностью компенсируется.
Почки - их функция состоит в удалении нелетучих кислот, главным образом серной кислоты. Почки должны удалять в сутки 40-60 ммоль ионов H+, накапливающихся за счет образования нелетучих кислот.
Их выделение происходит в проксимальных канальцах и собирательных трубках почек, где секретируются Н+, а в качестве буферных систем участвуют фосфаты, сульфаты (т.е. титруемые кислоты) и аммиак. Однако до того как может произойти экскреция всех кислот, почки должны реабсорбировать НСО3-, профильтровавшийся клубочками.
Способность канальцев почек к реабсорбции НСО3- высока. Самым важным местом реабсорбции НСО3- являются проксимальные канальцы, где посредством специального механизма происходит всасывание 90 % бикарбоната.
Угольная кислота образуется в клетке из воды и СО2 под действием карбоангидразы, Н+ активно переносятся через люминальную мембрану Na+/Н+-обменником (рис. 18.1.).
Рис. 18.1. Реабсорбция бикарбоната в клетках проксимального канальца. КА – карбоангидраза [по Шейману Д.А., 1999].
Затем НСО3- транспортируется через базолатеральную мембрану. Секретируемый Н +быстро соединяется с фильтруемым НСО3-, образуя угольную кислоту (Н2СО3). Угольная кислота превращается в воду и углекислый газ с помощью карбоангидразы (КА) на люминальной стороне щеточной каемки проксимального канальца. СО2 диффундирует обратно в клетку проксимального канальца, где соединяется с Н2О и образует угольную кислоту, завершая тем самым этот цикл.
Некарбоновые кислоты секретируются вставочными клетками собирательных трубок коры и наружного мозгового слоя почек (рис. 18.2.). Секреция Н+ в просвет канальцев происходит с помощью Н+-АТФазы, тогда как в реабсорбции НСО3- через базолатеральную мембрану участвует обменник Сl-/НСО3-.
Рис. 18.2. Секреция Н+ вставочными -клетками собирательной трубки. АДФ - аденозиндифосфат; АТФ – аденозинтрифосфат [по Шейману Д.А., 1999].
При защелачивании Н+ задерживается в клетках почек канальцев, а HCO-3 не реабсорбируясь выделяется.
Основная часть Н+ в моче должна быть выведена в форме буферов, обычно таких, как фосфаты и аммоний. Концентрация аммония регулируется преимущественно почками и колеблется в зависимости от, КОС. Объем суточной секреции кислот в наибольшей степени зависит от количества выделяемого аммония. NН4+ образуется и секретируется клетками проксимального канальца, а затем реабсорбируется в восходящем отделе петли Генле и концентрируется в мозговом слое почки. Небольшое количество NH4+ диссоциирует на NH3 и Н+, последний реабсорбируется. NH3 может диффундировать в собирательную трубку, где служит буфером для ионов Н+, секретируемых вставочными клетками.
В клетках почечных канальцев процесс образования аммиака (NH3) происходит также за счет дезаминирования аминокислот (глутаминовой), данный процесс показан на рис. 18.3.
Аммиак поступает в канальцевую мочу, где соединяясь с Н+ образует ионы аммония (NH4+), который присоединяя ионы Cl-, образует хлорид аммония и выводятся с мочой. Na+, освобожденной от Cl- в моче, всасывается в клетки почечных канальцев, соединяются с освобожденными от Н+ ионами HCO-3 и реабсорбируются в виде бикарбоната Na.
Рис. 18.3. Образование и секреция аммония клетками проксимального канальца. Глютамин переносится (на этом общем рисунке механизм не детализирован) в клетки через люминальную и базолатеральную мембраны и метаболизируется с образованием аммония и бикарбоната. Аммоний активно секретируется в просвет канальца (через Na, NH4+-антипорт), а бикарбонат переносится по градиенту через базолатеральную мембрану [по Вандеру А., 2000].
Желудочно-кишечный тракт. Клетки слизистой оболочки желудка секретируют НСl в очень высокой концентрации. При этом из крови ионы хлора поступают в полость желудка в соединении с Н+, образующимися в эпителии желудка при участии угольной ангидразы. Взамен хлоридов в плазму транспортируется бикарбонат. Существенного защелачивания крови при этом не происходит, т.к. ионы хлора желудочного сока достаточно быстро вновь всасываются в кровь в кишечнике. Железы слизистой оболочки кишечника секретируют щелочной сок, богатый бикарбонатами. При этом плазма пополняется Н+ в составе НС1. Кратковременный сдвиг реакции сразу же уравновешивается обратным всасыванием бикарбоната в кишечнике.
Печень участвует в поддержании кислотно-основного равновесия за счет удаления в основном кислых продуктов. В печени осуществляется синтез мочевины (СО(NH2)2) из шлаков, таких в частности, как аммиак (NH3) и хлорид аммония (NH4Cl), который поступает по портальной системе. Мочевина выводится почками. При острой и хронической почечной недостаточности синтез мочевины усиливается.
Ацидоз - это такое нарушение КОС, при котором в крови появляется относительный или абсолютный избыток кислот или недостаток оснований.
Алкалоз - это такое нарушение КОС, при котором имеется избыток оснований или недостаток кислот.
Классификация нарушений кислотно-основного равновесия
Ацидоз:
Газовый (дыхательный).
Негазовый.
метаболический
выделительный
экзогенный
комбинированный (напр., кетоацидоз + лактоацидоз; метаболический + выделительный; другие сочетания).
Смешанный (например, газовый + негазовый при асфиксии)
Алкалоз:
Газовый (дыхательный).
Негазовый.
выделительный
экзогенный
По степени компенсации все ацидозы и алкалозы подразделяются на
Компенсированные - это состояния, при которых в уравнении рН изменяются абсолютные количества угольной кислоты и натрия гидрокарбоната, но соотношение их остается 1:20. При этом рН существенно не изменяется, что служит показателем компенсации.
Декомпенсированные, когда изменяется не только общее количество угольной кислоты и натрия гидрокарбоната, но и их соотношение, о чем свидетельствует сдвиг рН крови за пределы нормы.
АЦИДОЗ
Газовый
снижение выведения СО2 при нарушениях внешнего дыхания (массивные пневмонии, ателектазы легких, бронхиальная астма, обструктивная форма эмфиземы легких, нарушения дыхания у ослабленных больных в раннем послеоперационном периоде, при синдроме трахео- бронхиальной непроходимости и т.д.).
высокая концентрация СО2 в окружающей среде.
При нарушении вентиляции легких основная компенсация дыхательного ацидоза осуществляется почками путем усиленного выведения Н+ и задержки (повышения реабсорбции) ионов НСО3- (в виде бикарбоната натрия). Такая компенсаторная реакция является целесообразной лишь до определенного момента. К выраженному респираторному ацидозу присоединяется второй патологический процесс - метаболический алкалоз.
Негазовый
кетоз (кетоацидоз) - сахарный диабет, голодание, нарушения функции печени, лихорадка, гипоксия и др.
лактатацидоз – тканевая гипоксия, нарушение функций печени, инфекции
обширные воспалительные процессы, ожоги, травмы
задержка кислот при почечной недостаточности (диффузный нефрит, уремия)
потеря щелочей - почечный канальцевый ацидоз, гипоксия, интоксикация сульфаниламидами, диарея, гиперсаливация
длительное употребление кислой пищи, прием кислот внутрь
Характерной компенсаторной реакцией при метаболическом ацидозе является дыхательный алкалоз.
Алкалоз
Газовый - усиленное выведение СО2 при гипервентиляции (неврозы, высотная болезнь, перед погружением у ныряльщиков, во время длительной операции или у реанимируемого больного).
При гипервентиляции развивается вазоконстрикция церебральных сосудов.
Компенсация дыхательного алкалоза осуществляется почками: задерживаются ионы Н+ и выделяется НСО3- . Кроме того, увеличивается количество органических кислот, в основном - молочной кислоты. Таким образом, все компенсаторные реакции являются целесообразными лишь относительно, так как приводят к возникновению метаболического ацидоза.
Негазовый.
потеря кислот - рвота при при декомпенсированном стенозе привратника, кишечная непроходимость, токсикоз беременных, гиперсекреция желудочного сока;
длительный прием щелочной пищи, стероиды, введение нитрата Na, альдостеронизм с гипокалийемией (приводит к избыточному выведению ионов Н+ почками и перемещению Н+ в клеточный сектор).
Компенсация метаболического алкалоза осуществляется за счет появления дыхательного ацидоза. Но такая компенсация приводит к раздражению дыхательного центра и гипервентиляции.
Часто недостаточное развитие компенсаторных реакций при метаболическом алкалозе объясняется еще и тем, что с одновременным защелачиванием плазмы внутри клеток развивается ацидоз. К+ усиленно выводится из клеток в плазму, сопряженно в клетки поступает Н+.
Развивается сложное нарушение КОС, характеризующееся внутриклеточным гипокалиемическим ацидозом и плазменным алкалазом.
Таблица 18.2.
Проявление нарушений кислотно-основного состояния.
Ацидоз |
Алкалоз |
|
|
51. – Этиология и патогенез эритроцитозов.
Эритроцитозы делятся на:
абсолютные (за счет увеличения синтеза )
-истинные
-симптоматические
относительные в результате
-эксикоза обезваживания
-перераспредилительные(стресс)
Абсолютный эритроцитоз отличается от относительного по ретикулоцитам
52. - Этиология и патогенез постгемораргической анемии
Анемии - это снижение концентрации НВ
Классификация причин:
1) кровопотеря
2) нарушение синтеза
3) повышенное разрушение
Классификация:
По размеру: 1а)микро- б)макро- в)нормо-
По цветному показателю эритроцитов: а)нормохромия б)гипохромия в)гиперхромия
По типу кроветворения: а)нормобластические б)мегалобластические
По способности к регенерации__________________
Особую группу составляют анемии со сложным многофакторным патогенезом, например: - у новорожденного; -В12 дефицит; анемия беременных и т.д.
Постгеморрагические анемии подразделяют на острые (острая потеря не менее 10% от ОЦК) и хронические
Стадии острой кровопотери:
рефлекторная (САР-> выброс эритроцитов из депо)-длится 4-5 часов
гидремическая (после 10 часов вода из межклеточного вещества выходит и срабатывает механизм РААС )
стадия усиленного кровитворения (в течении 3-5 дней -постепенный возврат к норме (ретикулоцитарный криз)).
Хроническая постгеморрагическая анемия. Основное звено патогенеза- дефицит Fe и гемическая гипоксия.
Этиология постепенное подкровливание где-либо (ЖКТ, матка, опухоли ).
Патогенез- симптомы со стороны ЦНС, глосситы, миоглобин понижается- мышечная слабость, сердечная слабость, кожа, кости
53. - Этиология и патогенез гемолитических анемий.
Анемии - это снижение концентрации НВ
Классификация причин:
1) кровопотеря
2) нарушение синтеза
3) повышенное разрушение
Классификация:
По размеру: 1а)микро- б)макро- в)нормо-
По цветному показателю эритроцитов: а)нормохромия б)гипохромия в)гиперхромия
По типу кроветворения: а)нормобластические б)мегалобластические
По способности к регенерации__________________
Особую группу составляют анемии со сложным многофакторным патогенезом, например: - у новорожденного; -В12 дефицит; анемия беременных и т.д.
Гемолиз бывает:
1) внутрисосудистый (укус, бледная поганка, малярия).
--гломерулы забиваются--------
--боль, почечная недостаточность
--озноб (пирогены)
2) внесосудистый (внутриклеточный- в синусах селезенки поедают макрофаги).
Гемолитические анемии делят на: 1)наследственные (гемоглобинопатии, серповидно-клеточная таласемия); 2) приобритенные.
Мембранопатии-дефект мембран (сфероцитоз, микроцитарная анемия Минковского-Шофара, овалоцитарная анемия).
Апаптоз- дефицит беталипопротеидов, участвующих в восстановительных реакциях (пируваткиназа, глю-6-фосфатдегидрогеназа).
Приобретенные гемалитические анемии. Гемолитические яды: анилин, мышьяк, бледная поганка, уксус.
54.–Дизэритропоэтические анемии. Этиология В12-дефицитной, фолиеводефецитной, железодефицитной и железорефрактерной анемии.
Гипохромная, микроцитарная анемия. Нормобластический тип кровотечения, регенерация, анизоцитоз, пойкилоцитоз. Вообще эта анемия относится к анемиям из-за нарушенного синтеза.Не только кровопотери, но и заболевания ЖКТ и печени могут привести к этой анемии. Fe – плохо усваемый элемент (может всасываться в двух видах:1) двухвалентное Fe; 2)в виде НВ ) для его усвоения необходимо ряд условий. Во первых наличие в пищи Fe-мясо, икра (у вегетарианцев всегда Fe-дефицитная анемия), затем HCl в желудке для ионизации, усваиваются ионы двухвалентного Fe. В тонком кишечнике Fe захватываются апоферритином энтероцитом в клетке (в виде феретина может немного депонироваться), затем трансферрин переносит в печень и красный костный мозг- синтез гемоглобин ( депо ------)
Могут быть нарушения на разных этапах: желудок…; кишечник…;печень…;
1-всасывание, 2-транспорт, 3-депо, 4-синтез.
Причем бывает, что железо не может включаться в гемм из-за отсутствия ферментов-это приводит к железорефрактерной, сидероакрестической анемии, когда нет Fe в эритроцитах, а в плазме много. Такая анемия бывает:
врожденная наследственная Х-рецесивная, не образует гемоглобин. Избыток Fe в направляется в печень –цирроз, надпочечники,яички-> сидероз органов
приобретенные: а) отравление свинцом-> блокируются ферменты Fe2+ пороририй; б) недостаток В6- нарушение включения Fe в гемм.
К анемиям вследствие нарушенного синтеза относится В12 и фолиеводефицитные анемии. Этиология: 1) Аутоиммунные повреждения обкладочных клеток (болезнь Аддисона - Бирмера); 2) Энтериты,--------; 3) Паразиты-лентец широкий.
Недостаток В12- нарушен синтез ДНК, т.к. необходим для синтеза тимина, это приводит к замедлению скорости образования ДНК и замедление деления быстро регенерирующих тканей: ЖКТ, кровь. Атрофический процесс в ЖКТ- лаковый язык, эритроциты долго созревают, большие т.к много в них НВ –Ц.П. повышается гиперхромная, мегалобластическая, мегалоцитарная анемия.
Нарушение образования миелина (коформа В12)- расстройство психики и локолюций.
Фолиеводефицитная анемия (мясо, дрожи, шпинат )-тоже нарушение репликации ДНК. Проявления те же, но без ЖКТ и нервной системы.
К анемиям в следствии нарушенного синтеза относятся гипопластические и апластические- нарушение эритропоэза (поврежденных или стволовых клеток).
Могут быть приобретенные из-за: 1) ионизирующего излучения; 2)лекарственных веществ(например: левомицитин) т.ж. угнетают белковый синтез цитостатики, яды, антифолиевые препараты: барбитураты, дифенин,нитрофураны,пирозолон.
Инфекции с эндоинтоксикацией. Опухоли- метастазы в кости. Вирусная инфекция-> аутоиммунный гемолиз. Печеночная и почечная недостаточность,NH3 оказывает миелотоксическое эффект.
55.- Лейкоцитозы и лейкопении. Виды. Этиология и патогенез. Основные типы изменения общего количества и отдельных видов лейкоцитов ворганизме. Типы ядерного сдвига.
Лейкоцитозы:
1) физиологические (у детей в первые дни, годы жизни проявляется лимфоцитоз, а так же при приеме пищи, физической работе и т. д.)
2) патологические из-за а) гемоконцентрации
б) перераспределительный (маргинальный 50%)
(адреналиновое встряхивание)
в) повышенный синтез клеток
Стимуляторы лейкопоэза:
Макрофагальные, лимфоцитарные- лейкопоэтины или колонийобразующие факторы (действуют начиная с миелоцитов)
Лейкоцитарные обломки (гной) и поэтины, трефоны нейтрофилов(лекарства, ядерные обломки, метилуроцил)
Бактериальные обломки, медиаторы и метаболиты своих и погибших клеток
Андрогены, глюкокортикоиды
Может быть отдельное увеличение форм лейкоцитов.
Нейтрофилез- микрофаги «дворники», причина: инфекция, асептическая гибель своих клеток (в результате травмы, миелолейкоза), конечно отразится и на общем числе лейкоцитов т. к. самая многочисленная популяция, их до 70%
Эозинофилия: глистные и аллергические реакции. Роль: выработка интерлейкинов, инактивация гистамина, разрежение воспаления - «алая, заря выздоровления», киллерное действие на многоклеточных- глистов
Базофилия тоже, но некоторые другие глисты (описторхоз) и аллергическая реакция чаще всего вместе с эозинофилами, т.к. базофилы и эозинофилы - ассоциация При хронических лейкозах опухолевой трансформации подвергаются бипотентные клетки, поэтому на периферию выходят из костного мозга базофилы и эозинофилы, иногда другие ростки (моноциты, тромбоциты и эритроциты при болезни Вакеза).
Лимфоцитоз при лимфолейкозе- опухоль лимфотического ростка. Хронические инфекции типа туберкулеза, бруцуллеза.
Моноцитоз: вирусные инфекции, простейшие, туберкулез (бульдозеры для убитых клеток входят в состав гранулемы).
Сдвиги лейкоцитарной формулы: влево и вправо.
Влево:
1) простой- увеличение палочкоядерных
2) регенераторный- появление метамиелоцитов
3) гиперрегенеративный- появление миелоцитов
4)гиперрегенератовно-дегенеративный сдвиг влево + дегенерации: кариопикноз, рексис, гиперсегментация более 5 сегментов, токсическая зернистость и вакуолизация цитоплазмы при тяжелых инфекциях- т.е. стимуляция с интоксикацией.
5) арегенераторный- исчезновение палочкоядерных, свидетельствующее об угнетения лейкопоэза. Возможны признаки дегенерации т. е. дегенеративный вправо.
Индекс сдвига – отношение несегментированных нейтрофилов к сегментированным (0,05-0,1).
Лейкимойдная реакция (ойдная – похожая на …) Может быть 50-300*10*9- такой лейкоцитоз, в основе которого- гиперэргическая реакция красного костного мозга- масса молодых форм до бластов в крови. Типы: меилойдного, лимфойдного, моноцитарного, лимфомоноцитарного, редко эозинофильного.
Лейкопении
Первичные- наследственные
Вторичные- приобретенные
п\г классификация напоминает таковую при анемиях.
вследствие уменьшенной продукции: алиментарная, токсическая, производные яды- бензол, лекарства: левомицетин, цитостатики, тяжелые инфекции, радиация, опухоли.
нарушение выхода из красного костного мозга: синдром «ленивых» лейкоцитов - наследственное понижение двигательной активности из-за дефекта мембран, уменьшения сократимости белков.
повышенное выделение из организма: свищи, ЖКТ-колиты, энтериты, плазмо- и лимфоррагии.
повышенное разрушение (аутоиммунные антитела к мембранам лейкоцитов, гаптеновые -осажденные лекарственные препараты на мембране лейкоцитов: сульфаниламиды, барбитураты, пиразолоны и выработка к ним антител).
перераспределительные: шоковые, коллаптойдные состояния из-за гемодилюции (гиперальдостеронизм, трансфузии)
Агранулоцитоз - резкое снижение гранулоцитов в крови (общее 1*10*9/л гранулоцитов соответственно 0,75*10*9/л)
Нарушение функции лейкоцитов при нормальном их количестве в крови (см. тему иммунодефициты).
===========================================================================
56. – Лейкозы. Этиология и патогенез. Лейкемеоидные реакции.
===========================================================================
Лейкозы
Гемобластозы - опухолевое поражение кроветворных клеток. Если опухолевые клетки диффузно и повсеместно поражают костный мозг- заболевание называют лейкозом. Раньше, для обозначения этого заболевания, применяли термин белокровие. Бывают красные лейкозы - болезнь Вакеза.
Этиология: почти тоже, что и у других опухолей.
1.Ионизирующая радиация
2.Химические канцерогены: циклические углеводороды, бензол и прочие составные бензина - антидетонаторы.
Лекарства: левомицетин, противовоспалительные - бутадион, индометицин (нарушен синтез простагландинов, уменьшение ЦАМФ), иммунодепрессанты.
3.Вирусная лимфома Беркитта- один из видов гематосарком.
Имеется наследственная предрасположенность к некоторым видам опухолей: казахи болеют лейкозами реже русских в одних местах проживания.
На фоне синдрома Дауна, Тернера, Кляйнфельтера (при которых наблюдается нестабильность генома) заболеваемость лейкозами возрастает в 20-30 раз.
Филадельфийская хромосома - делеция длинного плеча 22 пары хромосомы с транслокацией на 9 хромосому - почти 100% признак хронического миелолейкоза.
Патогенез такой же как и при других опухолях, - действительны генетические и эпигенетические теории возникновения. Те же 3 стадии опухолевой трансформации - индукция, промоция, прогрессия. Генетический анализ опухолевых клеток доказал моноклональное происхождение всех опухолевых клеток.На стадии опухолевой прогрессии - опухолевый рост приобретает поликлональный характер - из-за генетической нестабильности и естественного отбора более агрессивных клеток. Обязательно происходит смена более дифференцированных, зрелых, интенсивно размножающихся клеток менее зрелыми и более интенсивно размножающимися клетками. Если лейкоз был хроническим, то он переходит в острый и заканчивается бластным кризом.
Для лейкоза характерно:
1.Дедифференцировка, прекращение созревания
2.Клеточный атипизм (непохожие клетки (нарушено соотношение ядра и цитоплазмы))
3.Функциональный атипизм - лейкоциты не выполняют свою функцию.
4.Биохимический атипизм – опухолевые клетки - ловушки азота, глюкозы, витаминов; в них могут отсутствовать ферменты, типичные для этих клеток.
5.Метастазирование - мгновенное, учитывая особенности крови как жидкой ткани- инфильтраты в разных органах: кожа, почки, печень и т.д.
6.Автономность - не подчиняется регулирующим влияниям организма, вытеснение нормальных клеток своего и других ростков опухолевыми клетками.
Патогенез- вытеснение ростков- анемический синдром
кровоточивость (тромбоцитопении, лейкоинфильтрыты в печени - нарушения выработки факторов свертывания, повреждение сосудов)
септические осложнения (мало нормальных лейкоцитов, а лейкозные не выполняют свою функцию). Лейкоинфильтраты- симптоматика в зависимости от локализации, например, в ротовой полости язвенно – некротические поражения слизистой, осложняющиеся нагноением.
Токсическое влияние- на весь организм ( ловушки азота, глюкозы, нуклеопротеиды), много клеток гибнет, продолжительность жизни лейкозных клеток меньше, чем нормальных, обломки клеток
Механизмы угнетения других ростков крови:
Механическое сдавливание (вытеснение)
Фиброз (развитие стромы в костном мозге)
Конкуренция за питание
Токсическое действие продуктов метаболизма
Усиленный синтез кейлонов, которые не действуют на опухолевые клетки, но действуют на нормальные
Лимфолейкоз - синтез антител к другим клеткам
Классификация:
1)острый
2)хронический
Уровень клеток, которые подвергаются бластной трансформации- клетки 2,3,4 порядков
Опухолевая трансформация стволовой клетки - дедифференцированный лейкоз, если это происходит в миелобласте – миелолейкоз, лимфобласте – лимфолейкоз. Какой лейкоз острый или хронический зависит от степени дедифференцировки. Исключая случаи выздоровления, каждый хронический лейкоз переходит в острый, если он не умрет раньше от осложнений хронического.
Формы лейкоза в зависимости от ростка: миело-, лимфо-, моно-, плазмоцитарно-, мегакариоцитарно-, неклассифицируемые, редкие из лимфоцитарного ростка.
Болезнь Вальдстрема - усиленный синтез антител и называется:
при остром - миелобластный, лимфобластный
при хроническом - миелолейкоз, лимфолейкоз
при остром - бластный
при хроническом - весь спектр клеток, которые делятся и зрелые.
при остром - провал!!!
В зависимости от количества лейкоцитов в периферической крови различают:
1)лейкемический >50*10*9
2)сублейкемический > N <50*10*9
3)лейкопенический 4*10*9, но есть бласты
4)алейкимический <4*10*9 при некоторых видах лейкозах в перефирической крови нет бластов.
Отличия лейкозов от лейкемойдных реакций:
1)причина
2)лечение экс-ювантибус
3)анемия, тромбоцитопения, инфильтрация
4)базофильные и эозинофильные ассоциации при хроническом лейкозе.
5)филадельфийская хромосома при хроническом миелолейкозе
6)миелограмма в стерильных пунктатах - бластов 1-5% при реакции, а при лейкозах возрастает
7)онкобелки
8)хронический лимфолейкоз—Боткина-Гумпрехта. С острыми лейкозами не спутаешь, много бластов
Дифференцировка разных типов острых лейкозов трудна визуально, но хорошо при цитохимии.
Изменения в ротовой полости при лейкозах:
Болезнь Вакеза - «плетора» - кровоизлияния потехиальные на слизистых, носовые кровотечения, анемический синдром при острых лейкозах, бледность слизистых, кровоточивость десен, снижены тромбоциты.
Инфекционно-септические осложнения - некротические ангины, усугубление течений инфекций - дистрофические заболевания.
57. - Сердечная недостаточность. Этиология. Виды сердечной недостаточности. Механизмы компенсации при сердечной недостаточности.
Недостаточность кровообращения - патологическое состояние, обусловленное неспособностью организма обеспечить потребности органов и тканей в адекватном кровоснабжении.
Сердечная недостаточность (СН) - недостаточность кровообращения, обусловленная нарушением работы сердца.
Виды сердечной недостаточности
I. По первичности нарушения:
Первичная (кардиогенная) возникает в результате первичного снижения сократительной функции сердца при близкой к нормальной величине притока венозной крови к нему, т.е. развитие СН обусловлено первичным поражением сердца (например, миокардиты, пороки сердца).
Вторичная (некардиогенная) возникает в результате уменьшения венозного притока к сердцу при близкой к нормальной величине сократительной функции миокарда, т.е. развитие СН вторично и обусловлено другими заболеваниями сердечно-сосудистой и прочих систем (например, гипертоническая болезнь, тиреотоксикоз, коарктация аорты).
II. По происхождению:
Миокардиальная СН развивается при непосредственном поражении миокарда, когда, либо из функционирования выбывает участок сердечной мышцы, либо снижается сократительная функция миокарда вообще:
инфекционный и инфекционно-аллергический миокардит;
токсический миокардит;
гипоксическое и коронарогенное повреждение (последнее является причиной 60-70% СН у взрослых, в основном у мужчин);
дефицит субстратов;
кардиомиопатии (около 20%).
Перегрузочная СН возникает при перегрузке первично здорового миокарда, либо на начальных стадиях развития гипертонической болезни (когда миокард еще не изменен)
перегрузка объемом (изотоническая СН), встречается при клапанной недостаточности;
перегрузка давлением или сопротивлением (изометрическая СН) встречается при клапанных стенозах.
Смешанная СН (когда миокард и изменен, и перегружен) характерна для заключительных стадий гипертонической болезни, инфаркта папиллярной мышцы и некоторых других патологических процессов, в которых присутствуют оба компонента
III. По преимущественному поражению отдела сердца:
Левожелудочковая СН сопровождается застоем по малому кругу кровообращения, развивается вследствие митрального и аортального стеноза и/или недостаточности.
Правожелудочковая СН сопровождается застоем по большому кругу кровообращения, развивается вследствие
недостаточности и стеноза легочного ствола,
недостаточности и стеноза трикуспидального клапана,
дефекта межпредсердной и межжелудочковой перегородок,
триады и тетрады Фалло (стеноз легочного ствола, дефект межжелудочковой перегородки, декстрапозиция аорты, гипертрофия правого желудочка).
Тотальная СН.
IV. По скорости развития:
Острая СН характеризуется быстрым нарастанием клинических проявлений и может привести к смерти в короткие сроки (от нескольких минут до нескольких часов), например, вследствие отрыва хорды, инфаркта миокарда.
Хроническая СН характеризуется длительным течением с периодической сменой периодов обострения и компенсации (может тянуться годами), например, вследствие ишемической болезни сердца.
V. По степени выраженности хронической сердечной недостаточности
Начальная, скрытая.
Выраженная, с признаками недостаточности кровообращения в покое
а) слабовыраженные нарушения гемодинамики,
б) глубокие нарушения гемодинамики.
Конечная, дистрофическая.
Этиология сердечной недостаточности в основном изложена в представленной выше классификации. Дополнительно выделяют:
перикардиальную форму (при перикардитах),
эндокардиальную форму (обусловленную ревматическими пороками сердца),
сердечную недостаточность, вызванную врожденными пороками сердца.
Основное звено патогенеза сердечной недостаточности - снижение сократимости и насосной функции сердца.
В понятие «насосная функция сердца» включаются следующие показатели:
Сократимость – способность сердца сокращаться в ответ на возбуждение.
Конечно-систолический объем желудочка (или резервный объем) - количество крови, остающийся в желудочке в конце его систолы.
Конечно-диастолический объем желудочка - количество крови, находящееся в желудочке в конце его диастолы.
Ударный объем или систолический (УО иои СО) – количество крови, выбрасываемое из желудочка во время одного сокращения, оно равняется конечно-диастолическому объему желудочка минус конечно-систолический объем желудочка.
Минутный объем сердца (МОК) определяется количеством крови, выбрасываемым из каждого желудочка при каждом сокращении (ударный объем), умноженным на количество сокращений в минуту (частота сердечных сокращений).
Сердечный индекс (СИ) – отношение МОК к площади поверхности тела человека.
Сердечный выброс – объединенное название понятий МОК, СИ и УО.
Основные факторы, регулирующие насосную функцию сердца
Закон Франка-Старлинга – чем больше наполняется сердце во время диастолы, тем сильнее оно сокращается в систолу, т.е. ударный объем возрастает по мере увеличения наполнения сердца, следовательно, чем больше венозный возврат и растяжение волокон миокарда, тем больше сердечный выброс (рис. 21.3.).
Закон Анрепа – сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает.
Рефлекс Бейнбриджа - повышение давления в полых венах, правом предсердии приводит к рефлекторному увеличению частоты сердечных сокращений.
Лестница Боудича – чем выше частота сокращений сердца, тем (до определенного предела) выше сила его сокращений и наоборот.
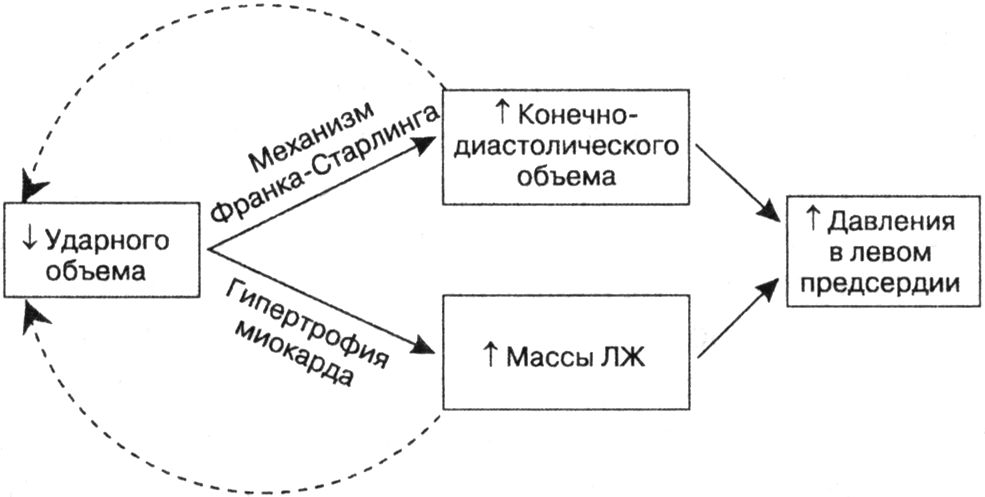
Рис. 21.3. Компенсаторные механизмы при сердечной недостаточности. Механизм Франка-Старлинга (включающийся при повышении конечно-дастолического объема ЛЖ) и гипертрофия миокарда (развивающаяся при перегрузке ЛЖ объемом или давлением) направлены на поддержание нормального ударного объема (пунктирная линия). Однако длительное повышение конечно-диастолического давления (за счет механизма Франка-Старлинга) и увеличение жесткости стенок ЛЖ (за счет гипертрофии миокарда) сопровождается повышением давления в левом предсердии, что нередко является причиной «обратной» сердечной недостаточности (например, застой в малом круге при левожелудочковой недостаточности) [по Лилли Л., 2003].
58. – Особенности патогенеза сердечной недостаточности при стенозе и недостаточности митрального и аортального клапана.
Схематическое изображение сердечных клапанов и камер сердца показано на рис. 21.18.
Рис. 21.18. Схематическое изображение сердечных клапанов и камер сердца [http://www.midatlanticcardio.com/html/termshtml/mitralvr.html]. Стрелками показано направление тока крови. The Valves of the heart – сердечные клапаны, Pulmonary Valve – клапан легочного ствола, Tricuspid Valve – трикуспидальный клапан, Aortic Valve – аортальный клапан.
Приобретенные пороки сердца
Наиболее частой причиной поражения клапанов и развития приобретенных пороков сердца является инфекция -гемолитическим стрептококком. Патогенез ревматического поражения клапанов показан на рис. 21.19., а морфологическая картина – на рис. 21.20.
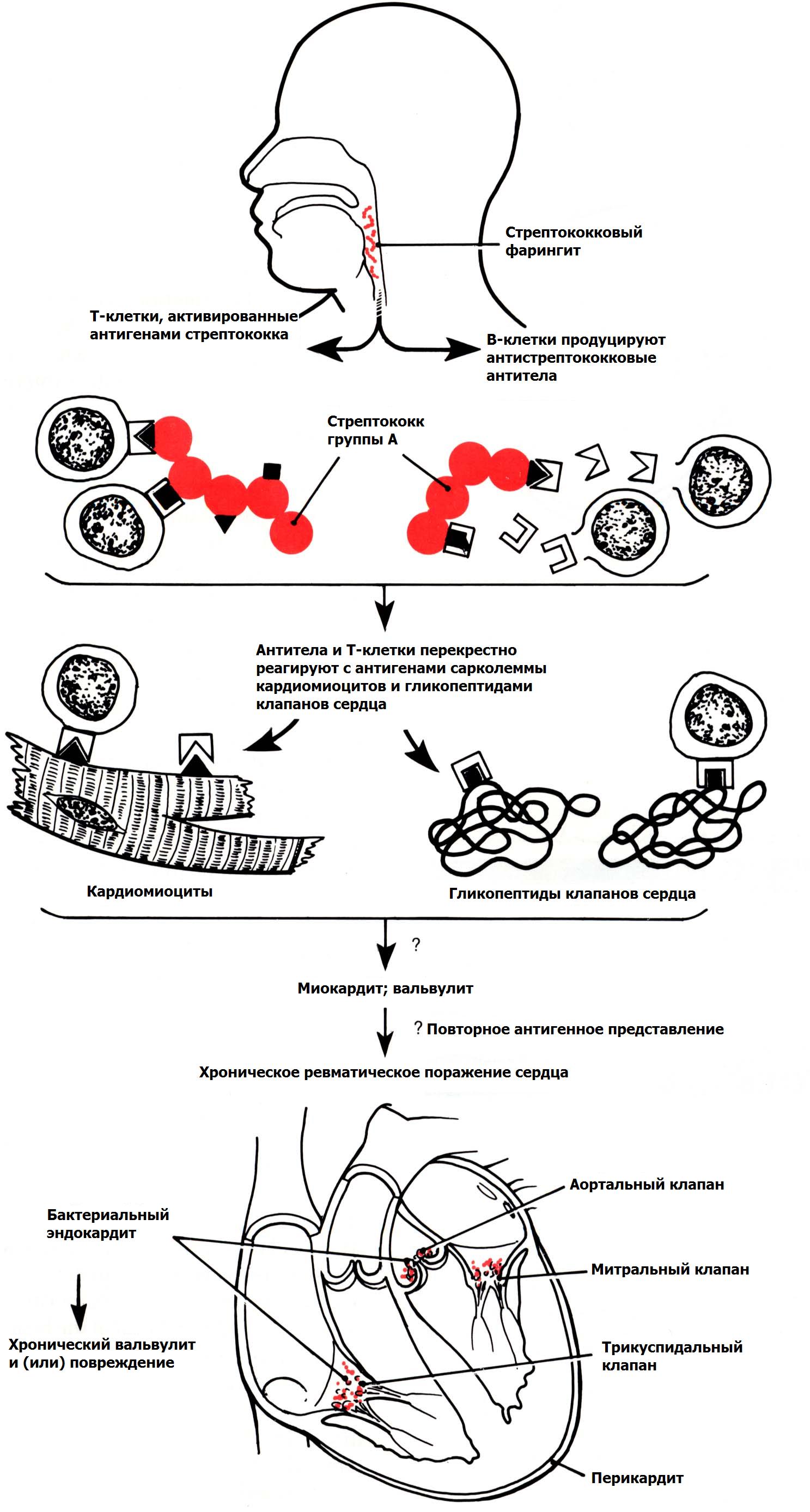
Рис. 21.19. Этиология и патогенез ревматического поражения сердца. [по Rubin E. and Farber J.L., 1994, с изменениями].
Рис. 21.20. Макропрепарат сердца при поражении бактериальным эндокардитом [http://www.pathology.vcu.edu/education/dental2/lab6.html]. Видны вегетации на створках митрального клапана.
Митральный стеноз. Препятствие оттоку крови из левого предсердия (ЛП) через суженный митральный клапан в левый желудочек увеличение давления в ЛП (перегрузка давлением) увеличение давления в легочных венах рефлекс Китаева (спазм легочных артериол при увеличении давления в легочных венах) увеличение сопротивления оттоку крови из правого желудочка гипертрофия левого предсердия и правого желудочка + гипертензия и застой крови по малому кругу отек легких.
Патофизиология митрального стеноза показана на рис. 21.21.
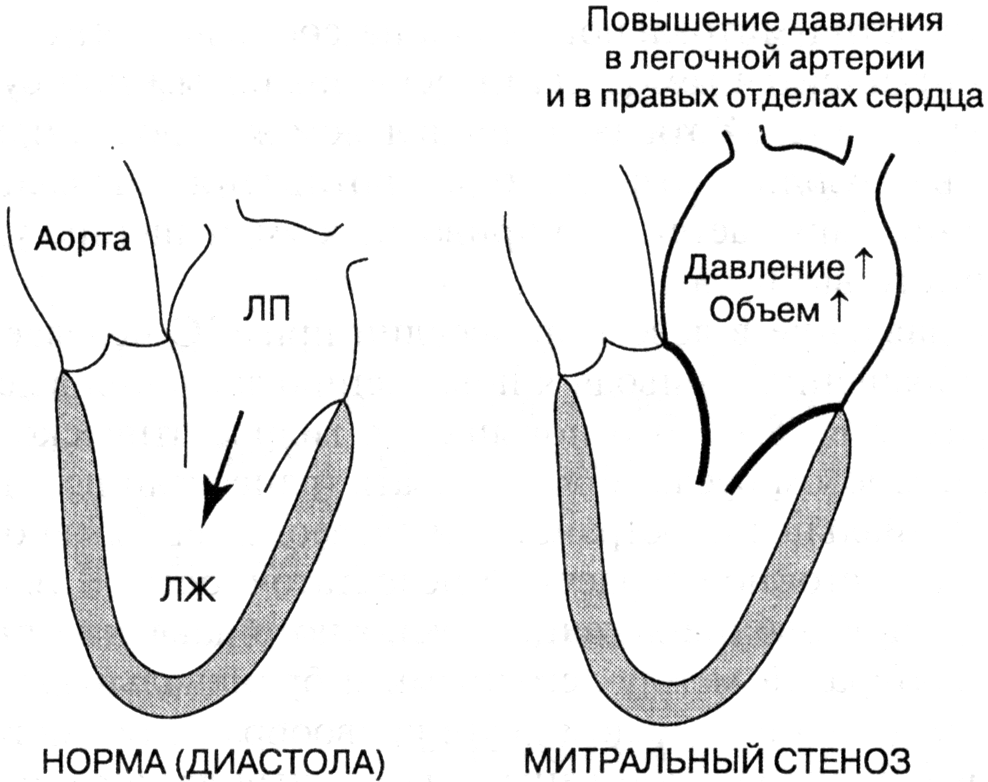
Рис. 21.21. Патофизиология митрального стеноза. В норме кровь в диастолу свободно течет из левого предсердия (ЛП) в левый желудочек (ЛЖ). При митральном стенозе имеется препятствие для опорожнения ЛП. Поэтому давление в ЛП увеличивается, что в свою очередь приводит к росту давления в малом круге кровообращения и в правых отделах сердца [по Лилли Л., 2003].
Митральная недостаточность. Неполное смыкание створок митрального клапана в систолу левого желудочка регургитация крови в ЛП (до 80%) увеличение объема крови в ЛП (перегрузка объемом) увеличение давления в легочных венах рефлекс Китаева (спазм легочных артериол при увеличении давления в легочных венах) увеличение сопротивления оттоку крови из правого желудочка гипертрофия левого предсердия и правого желудочка (меньше, чем при митральном стенозе) + гипертензия и застой крови по малому кругу отек легких. Учитывая, что большой объем крови из ЛП попадает затем в ЛЖ развивается еще и гипертрофия левого желудочка. При отрыве хорд развивается острая СН.
Патофизиология митральной недостаточности показана на рис. 21.22., изменения гемодинамики при проведении эходопплеркардиографии – на рис. 21.23.
Рис. 21.22. Патофизиология митральной недостаточности. В норме во время систолы сокращение левого желудочка проталкивает кровь только в аорту через аортальный клапан; закрытый митральный клапан препятствует регургитации в левое предсердие (ЛП). При митральной недостаточности (МН) часть выбрасываемой ЛЖ крови попадает ретроградно в ЛП, так что антеградный сердечный выброс в аорту снижается. При острой МН ЛП нормального размера и неэластичное, поэтому давление в нем значительно возрастает, и может развиться отек легких. При хронической МН ЛП увеличено и обладает большей податливостью, так что при нормальной систолической функции ЛЖ давление в нем ниже и признаки застоя в малом круге встречаются реже. Отмечается увеличение и эксцентрическая гипертрофия ЛЖ вследствие хронической перегрузки объемом [по Лилли Л., 2003].
Рис. 21.23. Обратный заброс крови в левое предсердие (регургитация) при митральной недостаточности [наблюдение Засорина С.В.].
Аортальный стеноз сопровождается препятствием оттоку крови из левого желудочка в аорту перегрузка давлением ЛЖ гипертрофия левого желудочка.
Патофизиология аортального стеноза показана на рис. 21.24.
Рис. 21.24. Патофизиология аортального стеноза (АС). Наличие препятствия на пути систолического тока крови из левого желудочка (ЛЖ) приводит к росту внутрижелудочкового давления и вторичной гипертрофии ЛЖ [по Лилли Л., 2003].
Аортальная недостаточность сопровождается регургитацией крови в ЛЖ из аорты перегрузка объемом ЛЖ гипертрофия левого желудочка.
Патофизиология аортальной недостаточности показана на рис. 21.25., изменения гемодинамики при проведении эходопплеркардиографии – на рис. 21.26.
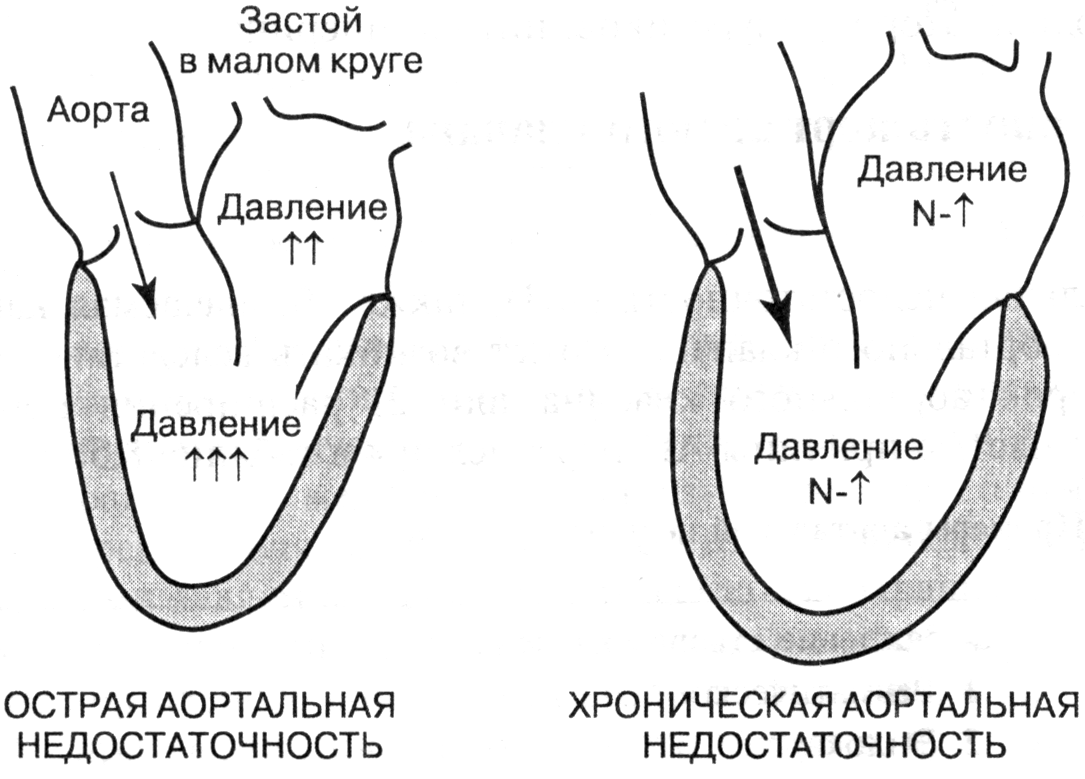
Рис. 21.25. Патофизиология острой и хронической аортальной недостаточности (АН). На каждой из схем показана патологическая регургитация крови из аорты в левый желудочек (большие стрелки). При острой АН левый желудочек имеет нормальные размеры и относительно низкую эластичность, так что диастолическое давление в нем значительно возрастает; оно передается на левое предсердие (ЛП) и легочное русло, приводя к застойным явлениям в легких или к их отеку. При хронической АН происходит компенсаторная дилатация ЛЖ и ЛП, так что больший объем регургитации вызывает меньшее повышение диастолического давления в ЛЖ, и застой в малом круге встречается реже. N - норма [по Лилли Л., 2003].
Рис. 21.26. Поток крови в систолу через аортальный клапан при его недостаточности (заброс крови в левый желудочек). Внизу – допплерограмма потока через клапан [наблюдение Засорина С.В.].
Рис. 21.24. Патофизиология аортального стеноза (АС). Наличие препятствия на пути систолического тока крови из левого желудочка (ЛЖ) приводит к росту внутрижелудочкового давления и вторичной гипертрофии ЛЖ [по Лилли Л., 2003].
Аортальная недостаточность сопровождается регургитацией крови в ЛЖ из аорты перегрузка объемом ЛЖ гипертрофия левого желудочка.
Патофизиология аортальной недостаточности показана на рис. 21.25., изменения гемодинамики при проведении эходопплеркардиографии – на рис. 21.26.
Рис. 21.25. Патофизиология острой и хронической аортальной недостаточности (АН). На каждой из схем показана патологическая регургитация крови из аорты в левый желудочек (большие стрелки). При острой АН левый желудочек имеет нормальные размеры и относительно низкую эластичность, так что диастолическое давление в нем значительно возрастает; оно передается на левое предсердие (ЛП) и легочное русло, приводя к застойным явлениям в легких или к их отеку. При хронической АН происходит компенсаторная дилатация ЛЖ и ЛП, так что больший объем регургитации вызывает меньшее повышение диастолического давления в ЛЖ, и застой в малом круге встречается реже. N - норма [по Лилли Л., 2003].
Рис. 21.26. Поток крови в систолу через аортальный клапан при его недостаточности (заброс крови в левый желудочек). Внизу – допплерограмма потока через клапан [наблюдение Засорина С.В.].
Трикуспидальная недостаточность. Неполное смыкание створок трикуспидального клапана в систолу правого желудочка регургитация крови в правое предсердие увеличение объема крови в правом предсердии и полых венах дилатация правого предсердия застой крови по большому кругу отеки на ногах + застойная венозная гиперемия печени сердечный цирроз печени.
Трикуспидальный стеноз (встречается редко) приводит к увеличению давления в ПЖ и в клинически выраженных случаях к снижению легочного кровообращения.
59. – Коронарная недостаточность. Виды. Этиология. Факторы риска ишемической болезни сердца.
Особенности коронарного кровообращения
Угол отхождения коронарных артерий от аорты близок к 90, что способствует турбуленции кровотока и тромбообразованию.
Затруднение тока крови по коронарным артериям в систолу из-за их компрессии (особенно ветвей левой коронарной артерии).
Поздние анастомозы между ветвями коронарных артерий, обуславливающие феномен обкрадывания при использовании спазмолитиков (чувствительными к препарату оказываются только «здоровые» венечные артерии, в то время как пораженные атеросклерозом артерии не расширяются, и основной поток крови перераспределяется в зоны миокарды, кровоснабжаемые непораженными сосудами).
Особенности метаболизма миокарда - основной источник энергии при окислительном фосфорилировании – жирные кислоты.
Регуляция коронарного кровообращения
Нервная.
Симпатическая иннервация: имеются преимущественно -адренорецепторы, но есть и -адренорецепторы; -адренорецепторы опосредуют увеличение ЧСС и одновременно расширяют коронарные артерии; при атеросклерозе нередко преобладают -эффекты (спазм).
Холинэргические влияния, вызывающие расширение коронарных артерий, выражены в меньшей степени.
Гуморальная («+» - сужение «-» - расширение).
Гормоны и биологически активные вещества |
Метаболиты |
|||||||||
Адреналин |
Ангиотеизин |
Тромбоксан |
Простациклин |
Гистамин |
Серотонин |
Брадикинин |
Тироксин |
СО2 |
Н+ |
Аденозин |
|
+ |
+ |
- |
- |
- |
- |
- |
- |
- |
- |
Абсолютная и относительная коронарная недостаточность. Этиология и патогенез ишемической болезни сердца и инфаркта миокарда.
Коронарная недостаточность - несоответствие между доставкой к сердцу кислорода и субстратов обмена веществ по коронарным артериям и потребностями миокарда в них.
Коронарная недостаточность – основная причина смерти в группе патологии сердечно-сосудистой системы, вместе с ишемической болезнью мозга составляет 90% смертности от сердечно-сосудистых заболеваний.
Абсолютная коронарная недостаточность - снижение доставки артериальной крови к миокарду по коронарным артериям, обусловленное уменьшением или полным закрытием просвета венечных артерий.
Причины, вызывающие абсолютную коронарную недостаточность, обозначают как коронарогенные (кардиальные):
Атеросклероз коронарных артерий, вызывающий их гемодинамически значимый стеноз и окклюзию – главная причина коронарной недостаточности.
Коронароспазм (спазм коронарных артерий), обусловленный повышением содержания катехоламинов, простагландинов (в основном тромбоксана А2) и увеличения адренореактивности сосудов миокарда (стенокардия Принцметалла). Также могут играть роль «миокардиальные мостики» - мышечные формирования, проходящие через миокард желудочков и покрывающие коронарные артерии.
Тромбоз и эмболия коронарных артерий (чаще как осложнение атеросклеротического их поражения).
Снижение коронарного резерва наблюдается при нарушениях регуляции коронарного кровотока, из-за потери эластичности сосудистой стенки, гипертрофии миокарда.
Коронарный резерв способен обеспечивать увеличение потребления О2 от 10 мл/мин/100 г в покое до 65 мл/мин/100 г вещества миокарда, т.е. коронарный резерв равен 55 мл/мин/100 г, а по кровотоку – в 4-5 раз.
Субинтимальные кровоизлияния в стенку коронарных артерий.
Снижение перфузионного давления в коронарных артериях при значительной бради- или тахикардии, трепетании (мерцании) предсердий или желудочков, недостаточности аортальных клапанов, острой гипотензии, сдавлении венечных сосудов опухолью, рубцом, инородным телом и т.п..
Относительная коронарная недостаточность - существенное повышение «запроса» и расхода миокардом кислорода и субстратов метаболизма по сравнению с уровнем их притока.
Причины, вызывающие относительную коронарную недостаточность, обозначают как некоронарогенные (экстракардиальные).
Стресс и активация симпато-адреналовой системы вызывает повышение потребности миокарда в кислороде вследствие возбуждения -адернорецепторов миокарда (что вызывает увеличение силы и частоты сердечных сокращений); у пациентов с имеющимся атеросклеротическим поражением коронарных артерий, кроме того, превалируют эффекты возбуждения -адренорецепторов коронарных артерий (спазм) над эффектами возбуждения -адренорецепторов.
Гипертония – срабатывает закон Анрепа (сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает), что повышает потребность миокарда в кислороде.
Тиреотоксикоз – избыток тиреоидных гормонов повышает адренореактивность миокарда и непосредственно увеличивает силу и частоту сердечных сокращений.
Анемия (острая постгеморрагическая) – резкое снижение кислородной емкости крови вызывает гемическую гипоксию и дефицит субстратов в миокарде.
60. – Патогенез инфаркта миокарда. Исходы инфаркта миокарда. Виды и стадии кардиогенного шока. Патогенез постинфарктного синдрома (Дресслера).
Инфаркт миокарда - возникновение некроза в сердце, обусловленное нарушением коронарного кровотока.
Различают крупноочаговый (трансмуральный) и мелкоочаговый (нетрансмуральный) инфаркт миокарда.
Патогенез ишемической болезни сердца и инфаркта миокарда обусловлен развитием стадийного процесса:
ишемия паранекроз некробиоз некроз и аутолиз исходы
При развитии инфаркта миокарда различают три зоны:
Ишемии – после восстановления кровоснабжения все изменения в ней подвергаются обратному развитию.
Ишемического повреждения (клетки в состоянии паранекроза и некробиоза) – после восстановления кровоснабжения часть этой зоны полностью восстанавливается, а часть – некротизируется.
Некроза – зона необратимых изменений, которая подвергается аутолизу, дальнейшая судьба может быть различной (см. «Исходы инфаркта миокарда»).
Исходы и осложнения инфаркта миокарда. Лабораторная и функциональная диагностика ишемической болезни сердца и инфаркта миокарда.
Исходы инфаркта миокарда
Рубцевание
Аневризма.
Разрыв и тампонада сердца.
Осложнения инфаркта миокарда
Аритмии.
Кардиогенный шок (пульсовое давление меньше 20 мм.рт.ст.)
Болевой шок – рефлекторное торможение (запредельное) центров сосудистой регуляции сосудистая недостаточность.
Истинный шок – обусловлен снижением насосной функции сердца из-за повреждения 50-65% миокарда острая сердечная недостаточность.
Аритмический шок - обусловлен снижением насосной функции сердца из-за некоординированных сердечных сокращений острая сердечная недостаточность.
Постинфарктный синдром Дресслера (триада: плеврит, аллергический пневмонит, перикардит) – развивается через 1 и более месяц после инфаркта миокарда, в основе лежит антигенное сходство некротизированных эндокардиальных клеток с клетками плевры, легкого и перикарда.
61. – Нарушение мозгового кровообращения. Виды. Этиалогия. Факторы риска ишемической болезни сердца.
Недостаточность мозгового кровообращения (сосудистая мозговая недостаточность) – несоответствие между энергетическими потребностями мозга, в первую очередь в кислороде, и его доставкой по артериям мозга.
Ишемическая болезнь мозга (ИБМ) – различные поражения мозга, обусловленные недостаточностью его кровоснабжения.
Инсульт – острое нарушение мозгового кровообращения со стойкой неврологической симптоматикой и морфологическими изменениями в веществе мозга.
Выделяют:
Ишемический инсульт – инфаркт мозга.
Геморрагический инсульт – кровоизлияние в мозг.
Классификация нарушений мозгового кровообращения
(по Шмидту Е.В. с сокращениями)
Начальные проявления недостаточности кровоснабжения головного мозга.
Преходящие нарушения мозгового кровообращения (ПНМК).
Транзиторные ишемические атаки (ТИА).
Гипертензивные церебральные кризы.
Инсульт.
Субарахноидальное нетравматическое кровоизлияние.
Геморрагический инсульт.
Ишемический инсульт (инфаркт).
Инсульт с восстановимым неврологическим дефицитом - малый инсульт.
Последствия ранее (более 1 года) перенесенного инсульта.
Прогрессирующие нарушения мозгового кровообращения.
Дисциркуляторная энцефалопатия (атеросклеротическая, гипертоническая).
Этиология нарушений мозгового кровообращения
Заболевания и патологические состояния, приводящие к нарушениям кровообращения в головном мозге
Атеросклероз.
Гипертоническая болезнь (эссенциальная гипертензия).
Болезни с симптоматической артериальной гипертензией.
Нейроциркуляторная дистония.
Артериальная гипотензия.
Поражения сердца и нарушения его деятельности.
Аномалии сердечно-сосудистой системы.
Поражение легких, ведущее к легочно-сердечной недостаточности с нарушением венозного кровообращения в головном мозге.
Инфекционные и аллергические васкулиты.
Токсические поражения сосудов мозга.
Заболевания эндокринной системы.
Травматические поражения сосудов мозга и его оболочек.
Сдавление артерий и вен (при изменениях позвоночника, опухолях и др.).
Болезни крови.
Характер поражения сосудов
Закупорка просвета сосуда
Сужение просвета сосуда (в %)
Перегибы, петлеобразование сосудов
Аневризмы:
мешотчатые;
артериовенозные;
каротидно-кавернозные, артериовенозные соустья;
Поражения вен и венозных синусов
Патогенетические механизмы ишемических поражений мозга
Обычно рассматриваются два основных механизма ишемических поражений – эмболия и сосудистая мозговая недостаточность. В первом случае ишемические поражения мозга называют эмболическими, а во втором – гемодинамическими. Развитие ишемических поражений мозга в большинстве случаев обусловлено эмболией, на долю же гемодинамических нарушений приходится 5-20% от общего числа всех ишемических поражений мозга. Хотя при окклюзирующих поражениях преобладает гемодинамический механизм.
Пороги ишемии мозга:
Функциональный (18-20 мл/100г/мин).
Деструктивных морфологических проявлений (5-10 мл/100г/мин).
Зона обратимых нарушений (10-23 мл/100г/мин), «ишемическая полутень» (penumbra): нейроны сохранены, но не функционируют, так как объемный мозговой кровоток в этой зоне достаточен для поддержания жизнеспособности нейронов, но недостаточен для их функционирования.
Стадии ишемии мозга (по Пауэрсу):
Стадия 0 (нормальное перфузионное давление): для обеспечения метаболических потребностей мозга регуляция мозгового кровообращения осуществляется за счет изменения просвета внутримозговых сосудов.
Стадия 1(небольшое снижение перфузионного давления): кровоток поддерживается за счет расширения сосудов, объем крови мозга при этом возрастает.
Стадия 2 (дальнейшее падение перфузионного давления): возможности вазодилатации исчерпываются, мозговой кровоток снижается, метаболизм поддерживается за счет повышения экстракции кислорода из артериальной крови.
При дальнейшем снижении перфузии мозга компенсаторные механизмы сосудистой системы оказываются несостоятельными, что приводит к развитию инфаркта мозга
Стадии ишемии мозга (по Бэрону):
Вазодилатация - снижение перфузионного давления компенсируется за счет увеличение объема крови в мозге.
Олигемия - мелкие внутричерепные артерии утрачивают способность к расширению, мозговой кровоток снижается; поддержание необходимого уровня потребления кислорода обеспечивается увеличением фракции извлечения кислорода, что получило название синдром «нищей перфузии».
Истинная ишемия - исчерпаны все возможные компенсаторные механизмы, уровень поглощения кислорода снижается, что может привести к развитию инфаркта мозга.
62. – Этиология и патогенез ишемических и геморрагических повреждений мозга. Патогенетические значения нахождения мозга в замкнутом пространстве черепной коробки.
63. – Сердечные аритмии. Этиология. Классификация.
Сердечные аритмии – нарушение частоты, ритмичности и последовательности сердечных сокращений.
Этиология аритмий: сердечные и внесердечные факторы.
Сердечные факторы.
Патология миокарда (миокардиты, кардимиоопатии, миокардиодистрофии).
Токсические и экзогенные воздействия на миокард.
Коронарная недостаточность (ишемическая болезнь сердца и инфаркт миокарда).
Клапанные пороки сердца.
Дополнительные проводящие пути в сердце.
Внесердечные факторы.
Нарушение вегетативной регуляции сердечной деятельности (например, повышение тонуса блуждающего нерва, при неврозах и психопатиях, нарушениях мозгового кровообращения).
Стресс (в первую очередь психоэмоциональный).
Повышение внутричерепного давления.
Гипо- и гипертиреоз.
Альдостеронизм (первичный и вторичный) вызывает гипокалийэмию.
Гипотермия и лихорадка.
Тромбоэмболия легочной артерии.
Гипо- и гипертония.
Болезнь и синдром Иценко-Кушинга (повышение секреции глюкокортикоидов, которые обладают слабым сродством к рецепторам альдостерона).
Передозировка некоторых лекарственных препаратов.
Аритмии, вызванные лекарственными препаратами
Часто причиной аритмий являются лекарственные препараты, обладающие собственной аритмогенной активностью. В первую очередь это относится к сердечным гликозидам и диуретикам.
Мочегонные препараты, усиливая экскрецию калия, способствуют возникновению гипокалиемии.
Сердечные гликозиды (дигиталис и др.) имеют свойство накапливаться в организме, ингибируя при этом Na+/К+-АТФазу, локализованную на сарколемме кардиомиоцитов. Снижение активности этого фермента сопровождается снижением уровня К+ и увеличением концентрации Na+ в саркоплазме. Накопление натрия в цитоплазме кардиомиоцитов приводит к усилению Nа+/Са2+-обмена, что сопровождается активным поступлением Са2+ в клетки миокарда и способствует усилению насосной функции сердца. Однако при этом формируется Са2+-перегрузка кардиомиоцитов. Кроме того, снижение внутриклеточной концентрации К+ вызывает замедление процессов реполяризации и тем самым способствует возникновению ранних деполяризаций и аритмий.
Лекарственные аритмии могут быть вызваны и антиаритмическими препаратами. У больных хронической сердечной недостаточностью, длительное время получавших блокаторы Nа+-каналов (флекаинид, этацизин и др.) или блокатор К+-каналов D-соталол, повышается частота случаев внезапной сердечной смерти.
D-соталол ингибирует К+-каналы, что ведет к замедлению процесса реполяризации, возникновению ранних реполяризации и опасных желудочковых аритмий.
Механизм аритмогенного действия блокаторов Nа+-каналов у пациентов с ХСН неизвестен.
Классификация аритмий. Аритмии, связанные с нарушением автоматии, возбудимости и проводимости.
Связанные с нарушением автоматии
А. Нарушения автоматизма синусового узла (номотопные аритмии)
Синусовая тахикардия
Синусовая брадикардия
Синусовая аритмия
Синдром слабости синусового узла
Б. Эктопические ритмы (гетеротопные аритмии)
Предсердный ритм
Узловой (атриовентрикулярный) ритм
Идиовентрикулярный (желудочковый) ритм
Миграция суправентрикулярного водителя ритма
Атриовентрикулярная диссоциация
Связанные с нарушением возбудимости
Экстрасистолия
Пароксизмальная тахикардия
Связанные с нарушением возбудимости и проводимости
Мерцание (фибрилляция) предсердий (мерцательная аритмия)
Трепетание предсердий
Трепетание и фибрилляция (мерцание) желудочков
Связанные с нарушением проводимости
Синоатриальная блокада
Внутрипредсердная блокада
Атриовентрикулярная блокада
Внутрижелудочковые блокады (блокады ветвей пучка Гиса).
Синдромы преждевременного возбуждения желудочков
а) Синдром Вольфа-Паркинсона-Уайта (WPW).
б) Синдром укороченного интервала PQ (CLC).
64. – Электрофизиологические механизмы аритмии: Изменения нормального автоматизма синоаурикулярного узла, аномальный автоматизм, постдеполяризация,блокады, реэнтри.
Фазы потенциала действия кардиомиоцита
Фаза 0. Во время этой начальной фазы возбуждения - фазы деполяризации - резко увеличивается проницаемость мембраны клетки для ионов Na+, которые быстро устремляются внутрь клетки (быстрый натриевый ток). При этом, естественно, меняется заряд мембраны: внутренняя поверхность мембраны становится положительной, а наружная - отрицательной. Величина ТМПД изменяется от -90 mV до +20 mV, т.е. происходит реверсия заряда - перезарядка мембраны. Продолжительность этой фазы не превышает 10 мс.
Фаза 1. Как только величина ТМПД достигнет примерно +20 mV, проницаемость мембраны для Na+ уменьшается, а для Cl- увеличивается. Это приводит к возникновению небольшого тока отрицательных ионов Cl- внутрь клетки, которые частично нейтрализуют избыток положительных ионов Na внутри клетки, что ведет к некоторому падению ТМПД примерно до 0 или ниже. Эта фаза носит название фазы начальной быстрой реполяризации.
Фаза 2. В течение этой фазы величина ТМПД поддерживается примерно на одном уровне, что приводит к формированию на кривой ТМПД своеобразного плато. Постоянный уровень величины ТМПД поддерживается при этом за счет медленного входящего тока Са2+ и Na+, направленного внутрь клетки, и тока К+ из клетки. Продолжительность этой фазы велика и составляет около 200 мс. В течение фазы 2 мышечная клетка остается в возбужденном состоянии, начало ее характеризуется деполяризацией, окончание - реполяризацией мембраны.
Фаза 3. К началу фазы 3 резко уменьшается проницаемость клеточной мембраны для Na+ и Са2+ и значительно возрастает проницаемость ее для К+. Поэтому вновь начинает преобладать перемещение ионов К+ наружу из клетки, что приводит к восстановлению прежней поляризации клеточной мембраны, имевшей место в состоянии покоя: наружная ее поверхность вновь оказывается заряженной положительно, а внутренняя поверхность - отрицательно. ТМПД достигает величины ТМПП. Эта фаза носит название фазы конечной быстрой реполяризации.
Фаза 4. Во время этой фазы ТМПД, называемой фазой диастолы, происходит восстановление исходной концентрации К+, Na+, Са2+, С1- соответственно внутри и вне клетки благодаря действию «Na+/K+-насоса». При этом уровень ТМПД мышечных клеток остается на уровне примерно -90 mV.
Клетки проводящей системы сердца и клетки синусового узла обладают способностью к спонтанному медленному увеличению ТМПП - уменьшению отрицательного заряда внутренней поверхности мембраны во время фазы 4. Этот процесс получил название спонтанной диастолической деполяризации и лежит в основе автоматической активности клеток синоатриального (синусового) узла и проводящей системы сердца, т.е. способности к «самопроизвольному» зарождению в них электрического импульса.
Основные электрофизиологические механизмы аритмий
Изменение нормального автоматизма сино-атриального узла
Изменение крутизны (скорости) спонтанной диастолической деполяризации: при ее возрастании пороговый потенциал возбуждения достигается быстрее и происходит учащение синусового ритма. Противоположный эффект, т.е. замедление спонтанной диастолическои деполяризации, ведет к замедлению синусового ритма.
Изменение величины мембранного потенциала покоя. Когда мембранный потенциал становится более отрицательным (при гиперполяризации клеточной мембраны, например при действии ацетилхолина), требуется больше времени для достижения порогового потенциала возбуждения (если скорость спонтанной диастолическои деполяризации остается неизменной). Следствием такого сдвига будет уменьшение числа сердечных сокращений. При снижении мембранного потенциала покоя ЧСС, напротив, возрастает.
Изменения критического уровня деполяризации (порогового потенциала возбуждения). Его уменьшение способствует учащению синусового ритма, а увеличение - брадикардии. Величина порогового потенциала возбуждения кардиомиоцитов определяется свойствами Nа+-каналов, а клеток проводящей системы - Са2+-каналов. В связи с этим следует напомнить, что в основе фазы быстрой деполяризации в клетках рабочего миокарда лежит активация быстрых Nа+-каналов, а в клетках проводящей ткани сердца - Са2+-каналов.
Аномальный (эктопический) автоматизм - это появление пейсмекерной активности в клетках сердца, не являющихся водителями сердечного ритма. Патологический автоматизм – образование участков эктопической активности в миокарде по механизму частичной деполяризации. Возникает в клетках при их частичной деполяризации (гипополяризация до 60-40 мВ). В норме эктопическая активность подавляется импульсами, поступающими из СА-узла, но при блокаде проведения импульса по предсердиям главным водителем ритма сердца может стать АВ-узел. Его способность к спонтанной деполяризации выражена менее, чем в клетках синусового узла, поэтому в условиях поперечной блокады обычно развивается брадикардия.
Еще менее выражена способность к автоматизму у волокон Пуркинье. Однако эти волокна, как и другие клетки проводящей системы, более устойчивы к гипоксии, чем сократительные кардиомиоциты, в связи с чем не всегда погибают в зоне ишемии. Вместе с тем электрофизиологические свойства таких ишемизированных волокон Пуркинье существенно отличаются от параметров интактных волокон тем, что у них появляется пейсмекерная активность, а способность к проведению импульса существенно снижается. Кроме того, спонтанная биоэлектрическая активность, возникающая в этих волокнах, в условиях патологии (например, при глубокой ишемии) перестает подавляться импульсами, поступающими из синусового узла, и может быть причиной возникновения желудочковых экстрасистол.
Постдеполяризация и триггерная активность. Постдеполяризация – вторичные подпороговые деполяризации, которые могут появляться:
а) во время 2-3 фаз ПД – ранние постдеполяризации.
Ранняя постдеполяризация - это преждевременная деполяризация клеток миокарда и проводящей системы, которая появляется тогда, когда фаза реполяризации потенциала действия еще не завершена, потенциал мембраны еще не достиг потенциала покоя. Этот преждевременный ПД рассматривается как триггерный (наведенный), поскольку он обязан своим возникновением ранней пост деполяризации, исходящей от основного ПД. В свою очередь, второй (наведенный) ПД за счет своей ранней постдеполяризации может вызвать третий, тоже триггерный ПД, а третий ПД - четвертый триггерный ПД и т.д. Если источник триггерной активности находится в желудочках, то на ЭКГ подобный тип нарушений образования импульсов проявляется, как желудочковая экстрасистолия или полиморфная желудочковая тахикардия.
Можно указать таких два важнейших условия возникновения ранних постдеполяризаций, как: удлинение фазы реполяризации потенциала действия и брадикардия. При замедлении реполяризации и, соответственно, увеличении общей продолжительности ПД может возникнуть преждевременная спонтанная деполяризация в тот момент, когда процесс реполяризации еще не завершился. При уменьшении частоты основного ритма сердца (брадикардия) происходит постепенное возрастание амплитуды ранних постдеполяризаций. Достигнув порога возбуждения, одна из них вызывает образование нового ПД еще до завершения исходного.
Поскольку ранние постдеполяризации реализуются за счет активации Na+- и Са2+-каналов, супрессировать связанные с ними нарушения сердечного ритма можно с помощью блокаторов названных каналов.
Возникновению ранних постдеполяризаций способствуют: гиперкатехоламинемия, гипокалиемия, ацидоз, ишемия, синдром удлиненного интервала Q-T. Часто подобный автоматизм является результатом применения антиаритмических препаратов, блокирующих К+-каналы (соталол, хинидин и др.).
б) непосредственно после ПД – задержанные (поздние) постдеполяризации
Поздние (задержанные) постдеполяризации - это преждевременная деполяризация клеток миокарда и проводящей ткани, которая появляется сразу же после завершения фазы реполяризации. Возникают, как правило, после частичной гиперполяризации (следовые потенциалы). Если амплитуда постдеполяризации достигает КУД, возникает ПД и т.д. Подпороговые колебания мембранного потенциала, которые в норме могут присутствовать, но никогда себя не проявляют, при патологических состояниях, вызывающих Са2+-перегрузку кардиомиоцитов, могут возрастать по амплитуде, достигая порога возбуждения.
Повышение внутриклеточной концентрации ионов кальция вызывает активацию неселективных ионных каналов, обеспечивающих усиленное поступление катионов из внеклеточной среды в кардиомиоцит. При этом в клетку поступают главным образом ионы Na+, концентрация которых в экстрацеллюлярной жидкости намного превышает уровень К+ и Са2+. В результате отрицательный заряд внутренней поверхности клеточной мембраны уменьшается, достигая пороговой величины, вслед за чем возникает серия преждевременных ПД. В конечном итоге формируется цепь триггерных возбуждений.
Триггерная активность клеток сердца, связанная с задержанными постдеполяризациями, может возникнуть под действием сердечных гликозидов или катехоламинов. Очень часто она появляется при инфаркте миокарда.
Рис. Б – ранние постдеполяризации; В – поздние постдеполяризации.
Нарушение проводимости (блокады)
Снижение потенциала покоя (например, в зоне ишемии инфаркта миокарда) сопровождается замедлением нулевой фазы ПД, т.к. уровень ПП коррелирует с процентом открытых быстрых Nа+-каналов в мембране. При снижении ПП до 50 мВ (в норме 80-90 мВ) инактивируется около 50% Nа+-каналов и возбуждение (проведение) становится невозможным (блокада).
Декрементное (затухающее) проведение происходит, вероятно, под влиянием увеличения внеклеточной концентрации К+, например, после инфаркта миокарда.
Неравномерное проведение – в параллельных волокнах скорость изменяется неравномерно, деполяризация происходит неравномерно и общая эффективность стимула падает (частичная или полная блокада).
Повторный вход импульса (re-entry) - явление, при котором импульс, совершающий движение по замкнутому кругу (петле, кольцу), возвращается к месту своего возникновения, совершая круговое движение.
Различают macro re-entry (макрореентри) и micro re-entry (микрореентри). При таком делении учитывают размеры петли (круга), в которой осуществляется повторный вход.
Для формирования macro re-entry с характерными для него свойствами требуются определенные условия:
наличие устойчивой замкнутой петли, длина ее зависит от анатомического периметра невозбудимого препятствия, вокруг которого движется импульс;
однонаправленная блокада проведения в одном из сегментов петли re-entry;
продолжительность распространения волны возбуждения должна быть короче времени, за которое импульс может преодолеть всю длину петли re-entry. Благодаря этому перед фронтом распространяющегося по кругу импульса имеется участок ткани, вышедший из состояния рефрактерности и успевший восстановить свою возбудимость («окно возбудимости»).
Механизм macro reentry лежит, как полагают, в основе трепетания предсердий.
Устранить подобную циркуляцию можно с помощью удлинения периода рефрактерности. При этом «окно возбудимости» может закрыться, поскольку циркулирующая волна наталкивается на участок, находящийся в состоянии рефрактерности. Добиться этого можно с помощью антиаритмических препаратов, блокирующих К+-каналы, что ведет к замедлению реполяризации и увеличению продолжительности рефрактерного периода. В этом случае «окно возбудимости» закрывается, и движение импульса прекращается.
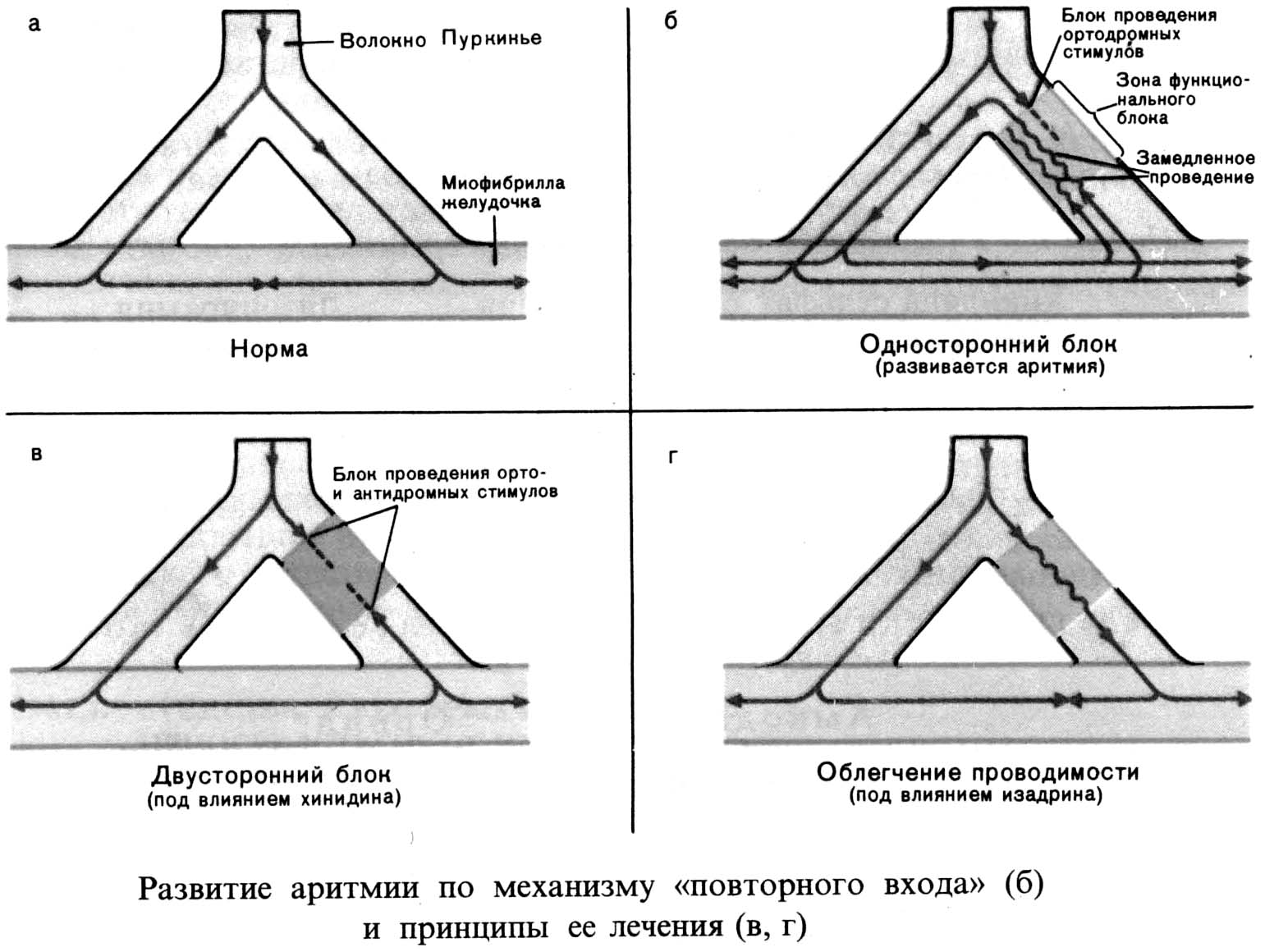
При micro re-entry движение импульса происходит по малому замкнутому кольцу, не связанному с каким-либо анатомическим препятствием. Импульс совершает не только круговое, но и центростремительное движение. Ближе к центру ПД снижается, и возбуждение затухает, клетки в центре дают только локальный ответ, т.к. находятся в состоянии рефрактерности и как бы заменяют анатомическое препятствие.
По-видимому, многие сложные тахиаритмии, в частности фибрилляции, связаны с механизмом micro re-entry. Сочетания петель, лежащих в разных плоскостях, возникают у больных с желудочковыми тахикардиями в остром периоде инфаркта миокарда.
Очень часто морфологическим субстратом для возникновения re-entry являются волокна Пуркинье, находящиеся в зоне ишемии. Эти клетки устойчивы к гипоксии и могут не погибать в очаге инфаркта. Однако при этом они меняют свои электрофизиологические характеристики таким образом, что быстрые Na+-каналы превращаются в «медленные». В этом случае проведение импульса замедляется и из зоны ишемии он выходит в тот момент, когда остальной миокард уже находится в состоянии относительной рефрактерности и готов к повторному возбуждению, но импульс из синусового узла еще не поступил. Возникает феномен повторного входа (re-entry), когда миокард дважды стимулируется одним и тем же импульсом: первый раз, когда он поступает из синусового узла, и второй раз, когда он повторно выходит из зоны ишемии. В этом случае разорвать петлю re-entry можно с помощью препаратов, блокирующих «медленные» Nа+-каналы в зоне ишемии (лидокаин, новокаинамид).
Несомненным достоинством этих антиаритмиков является то, что они проявляют высокое сродство именно к аномальным Nа+-каналам в зоне ишемии и практически не ингибируют быстрые Na+-каналы в клетках здорового миокарда, а значит, не влияют на электрофизиологические процессы в интактных кардиомиоцитах.
Отраженный повторный вход (reslected re-entry) - основе электротоническое проведение через зону функциональной невозбудимости в анте- и ретроградном направлении с повторным возбуждением проксимального волокна, вышедшего из состояния рефрактерности.
65. – Виды нарушений артериального давления. Артериальные гипертензии. Этиология. Осложнения.
Артериальная гипертония (гипертензия) - стойкое повышение АД.
Артериальная гипотония (гипотензия) - стойкое понижение АД.
Термины «гипер-» и «гипотония» применяются для обозначения в большей степени сосудистого тонуса, а термины «гипер-» и «гипотензия» - для обозначения артериального давления.
Гипертоническая болезнь (эссенциальная гипертония, первичная артериальная гипертензия) - заболевание, характеризующееся стойким (первичным) повышением АД ("неизвестной природы") при отсутствии патологических изменений какого-либо органа, устранение которых может привести к его нормализации.
Классификация артериальных гипертоний по величине ад у детей и у взрослых
Формулы на расчеты величины артериального давления у детей
|
Систолическое |
Диастолическое |
Среднее возрастное |
90+2n |
60+n |
Верхнее пограничное |
105+2n |
75+n |
Нижнее пограничное |
75+2n |
45+n |
n – возраст в годах
Артериальное давление (в мм рт.ст) (М.Я. Студеникин, 1981)
Возраст |
Средняя величина и пределы колебания |
|
Максимальное |
Минимальное |
|
1-3 года |
114 (85 – 120) |
58 (40 – 80) |
4-7 лет |
103 (85 – 120) |
56 (35 – 70) |
8-12 лет |
123 (95 – 140) |
60 (45 – 80) |
13-16 лет (мальчики) |
113 (95 – 140) |
60 (35 – 90) |
13-16 лет (девочки) |
118 (100 – 150) |
62 (40 – 85) |
Артериальное давление и артериальные гипертензии
Характеристика |
АД, мм.рт.ст. |
Типовые клинические проявления |
Нижняя граница нормы |
100/60 |
|
Верхняя граница нормы |
139/89 |
|
Артериальная гипертония |
|
|
Пограничная |
140-159/90-94 |
Функциональные. |
I стадия (легкая) |
160-179/95-104 |
|
II стадия (среднетяжелая) |
180-200/105-114 |
Гипертрофия левого желудочка, ангиоретинопатия, НМК. |
III стадия (тяжелая) |
> 200/115 и > |
Склеротические изменения. |
Артериальная гипотония |
< 100/60 |
НМК, обморок, коллапс, шок. |
Классификация АД ВОЗ (JNC, 1999 г.)
ВОЗ – Всемирная Организация Здравоохранения; JNC (Joint National Committee on Prevention, Detection, Evaluation and Treatment of High Blood Pressure) – Объединенный Национальный Комитет по профилактике, диагностике, оценке и лечению высокого артериального давления.
|
АД, мм рт. ст. |
|
систолическое |
диастолическое |
|
Оптимальное АД |
< 120 |
<80 |
Нормальное АД |
< 130 |
<85 |
Повышенное нормальное АД |
130-139 |
85-89 |
Артериальная гипертензия |
||
1-я степень ("мягкая") |
140-159 |
90-99 |
Подгруппа: пограничная |
140-149 |
90-94 |
2-я степень ("умеренная") |
160-179 |
100-109 |
3-я степень ("тяжелая") |
> 180 |
> 110 |
Изолированная систолическая гипертензия |
>140 |
< 90 |
Подгруппа: пограничная |
140-149 |
< 90 |
* Если показатели систолического и диастолического АД находятся в разных классах, уровень АД у данного больного относят к более высокому классу.
В классификации ВОЗ-JNC от 1999 г. 1, 2 и 3-я степени артериальной гипертензии соответствуют терминам "мягкая", "умеренная" и "тяжелая" гипертензия, которые использовались, например, в рекомендациях ВОЗ-МОГ 1993 г.
Классификация нарушений ад
Преходящие
гипертензивные реакции,
гипотензивные реакции.
Острые
гипертонический криз,
гипотонии.
Стойкие
Артериальные гипертонии
Первичная (эссенциальная) артериальная гипертония.
Вторичные (симптоматические) артериальные гипертонии.
почечные (ренопривная и вазоренальная или реноваскулярная),
эндокринные (надпочечниковые: минералокортикоидные, глюкокортикоидные, катехоламиновые, гипертиреоидные),
нейрогенные,
гемодинамические.
Артериальные гипотонии
Физиологическая артериальная гипотония.
конституциональная,
адаптивная (тренированность, высокогорье).
Нейроциркуляторная (первичная) гипотония.
Симптоматические (вторичные) гипотонии (при язвенной болезни желудка и двенадцатиперстной кишки, гепатите, пневмонии и туберкулезе, интоксикации, ревматоидном артрите, остеохондрозе шейного отдела позвоночника).
66. – Современные представления об этиологии и патогенезе гипертонической болезни.
Теории развития гипертонической болезни (эссенциальной гипертензии)
Гипотеза Э. Геллъгорна и соавт.
Инициальным патогенетическим фактором является стойкая повышенная возбудимость и реактивность (гиперергия) высших симпатических нервных центров (в частности, расположенных в заднем отделе гипоталамуса). Факторами, вызывающими стойкую гиперергию этих центров, является длительное, повторное возбуждение эмоциональных центров, тесно связанных с симпатическими ядрами гипоталамуса.
Гиперергическое состояние указанных структур вызывает, с одной стороны, повышение тонуса прессорных центров (по природе своей являющихся симпатическими), что ведет к спазму сосудов, увеличению сердечного выброса и повышению АД, а с другой стороны, обусловливает гиперпродукцию гуморальных факторов с прессорным действием адреналина, норадреналина, вазопрессина, АКТГ, кортикостероидов, а также гиперсекрецию ренина в юкстагломерулярном аппарате (ЮГА) почек.
Гипотеза е. Муирад, а. Гайтона и соавт.
Инициальным фактором развития гипертензии является генетически обусловленный пониженный уровень NaCl- и водовыделительной функции почек. Это ведет к накоплению избытка натрия и воды в организме, включая ткань сосудистых стенок, в том числе их гладкие мышцы. В связи с этим развивается гиперволемия, повышаются тонус сосудов и чувствительность их стенок к прессорным гормонам и другим биологически активным факторам.
Гипотеза нарушения функций мембранных ионных насосов проф. Ю.В. Постнова
Инициальным фактором в патогенезе артериальной гипертензии является генерализованный наследственный дефект мембранных ионных насосов клеток, включая гладкомышечные клетки стенок артериол. Дефект этот заключается в снижении активности кальциевого насоса, локализующегося в мембранах эндоплазматической сети, а также - натриевого насоса, локализующегося в плазмолемме. В результате уменьшается, с одной стороны, «откачивание» Ca+2 из цитоплазмы в эндоплазматическую сесть, а с другой стороны, к уменьшению «откачивания» Na+ из цитоплазмы в межклеточное пространство. Избыток Ca+2 и Na+ в цитоплазме гладкомышечных клеток сосудов вызывает их спазм, а также повышение чувствительности к прессорным факторам.
Гипотеза проф. Г.Ф. Ланга и проф. А.Л. Мясникова
Инициальным патогенетическим фактором развития ГБ является растормаживание центров CCС (снижение тормозного влияния коры головного мозга, оказываемого ею в норме на подкорковые вегетативные нервные центры, прежде всего - на прессорные). Это ведет, с одной стороны, к спазму артериол и повышению АД, а с другой - к обусловленному спазмом почечных артерий и другими изменениями включению почечного прессорного патогенетического фактора, эндокринных и рефлексогенного механизмов повышения АД. Причиной снижения тормозного влияния коры головного мозга на подкорковые прессорные центры (что ведет к развитию гипертонической болезни) является ослабление ее «тонуса» (активности) под влиянием избыточных сигналов, поступающих от экстеро- и интерорецепторов (стрессоры).
Этиология – стрессоры (чаще всего психические).
Первичное звено – возбуждение симпатических центров включение САР, подключение ЮГА почек альдостерон накопление Na+ увеличение ОЦК + набухание сосудистой стенки повышение ее реактивности.
Длительный спазм гладких мышц сосудов накопление Ca+2 в цитоплазме мышечных волокон (механизм «bodybuilding») активация РНК-полимеразы гипертрофия гладкомышечных элементов сосудистой стенки.
Факторы риска гипертонической болезни
Неконтролируемые факторы риска.
Наследственность.
Контролируемые факторы риска.
Избыточная масса тела (увеличение массы тела увеличение ОЦК по закону Франка-Старлинга и закону Анрепа увеличивается сила сердечных сокращений АД). Нарушение чувствительности к инсулину ангиопатии.
Гиподинамия (сосуды мышц содержат -адренорецепторы + при работе накопление аденозина, СО2, Н+).
Употребление большого количества NaCl (лица с низкой сольчувствительностью).
Курение и алкоголь.
Сахарный диабет.
Атеросклероз (приводит к поражению депрессорных зон аорты): адреналин усиление липолиза накопление ЖК накопление холестерина.
Клинико-потегенетические формы эссенциальной гипертонии по Кушаковскому
Гиперадренерическая (Ланг, Мясников).
Гипергидратационная.
Ангиотензин–зависимая.
Кальций–зависимая (Постнов, Орлов).
Цереброишемическая.
Порочные круги при гипертонической болезни
Рениновый: АД артериальный спазм ишемия почек выброс ренина из ЮГА активация РААС АД.
Сердечный: гиперволемия закон Анрепа (сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает) рост УО АД.
Атеросклероз поражение депрессорных зон.
Сосудистый: спазм гладких мышц сосудов накопление Ca+2 в цитоплазме мышечных волокон активация РНК-полимеразы гипертрофия и гиперплазия гладкомышечных элементов сосудистой стенки. периферического сосудистого сопротивления.
Осложнения артериальных гипертензий
Гипертонический криз - одно из наиболее частых и тяжелых осложнений гипертонической болезни и симптоматических артериальных гипертензий, характеризующееся острым повышением артериального давления до индивидуально высоких цифр и резким обострением симптоматики заболевания с преимущественным преобладанием церебральных и сердечно сосудистых расстройств.
Вторичная (некардиогенная) сердечная недостаточность, гипертрофия миокарда, инфаркт миокарда.
Инсульт (ишемический и геморрагический).
Первично-сморщенная почка (артериологиалиноз и артериолосклероз почечных сосудов нарушение почечной гемодинамики структурные изменения в нефронах и интерстициальной ткани постепенно нарастающая гибель почечных нефронов, сопровождающаяся уменьшением массы действующих нефронов), клиническим проявлением которой является хроническая почечная недостаточность (никтурия, полиурия, понижение относительной плотности мочи, азотемия).
Ангиоретинопатия.
67. – Особенности этиологии и патогенеза различных видов симптоматических артериальных гипергензий.
Симптоматические гипертензии. Патогенетическая классификация. Осложнения артериальных гипертензий.
Симптоматические (вторичные) артериальные гипертонии (гипертензии) - стойкое повышение АД, являющееся проявлением (симптомом) какого-либо заболевания.
Почечные артериальные гипертензии
Вазоренальная: атеросклероз почечной артерии, фибромускулярная дисплазия, аортоартериит, васкулиты, эндартериит, тромбоз, эмболия, аневризмы почечной артерии, атрезия и гипоплазия почечных артерий, артериовенозные фистулы, стенозы и тромбозы вен, гематомы, травмы сосудов почек; неоплазмы, сдавливающие почечные артерии ишемия почек выброс ренина из ЮГА активация РААС АД.
Ренопривная: поражение (гломерулонефрит, хронический пиелонефрит, диабетический гломерулосклероз, амилоидоз, гидронефроз) или удаление паренхимы почек дефицит почечных простагландинов А и Е) АД.
Эндокринные артериальные гипертензии
Минералокортикоидные (первичный альдостеронизм или синдром Конна): гиперплазия или опухоль клубочковой зоны коры надпочечников повышение продукции альдостерона усиление реабсорбции Na+ и воды увеличение концентрации Na+ в плазме гиперосмия плазмы стимуляция секреции вазопрессина рост ОЦК АД.
увеличение концентрации Na+ в плазме повышение транспорта ионов Na+ через мембраны клеток накопление избытка Na+ в клетках набухание клеток, в том числе и стенок сосудов сужение их просвета + увеличение чувствительности к катехоламинам, вазопрессину, АГII АД.
Глюкокортикоидные: гиперплазия или гормонально-активные опухоли пучковой зоны коры надпочечников (синдром Иценко-Кушинга) и базофильных клеток передней доли гипофиза (болезнь Иценко-Кушинга) пермиссивное действие глюкокортикоидов на эффект адреналина + слабая минералокортикоидная активность глюкокортикоидов АД.
Катехоламиновые: феохромоцитома (опухоль мозгового вещества надпочечников) гиперпродукция адреналина и норадреналина АД.
Гипертиреоидные: тиреотоксикоз гипертиреоидный зоб щитовидной железы и т.п. гиперпродукция тироксина и трийодтиронина увеличение силы и частоты сердечных сокращений увеличение УО АД.
гиперпродукция тироксина и трийодтиронина увеличение чувствительности сердца к катехоламинам АД.
Вазопрессиновые: повышение секреции гипоталамусом вазопрессина (вазопрессинома) сужение сосудов + задержка воды с увеличением ОЦК АД.
Нейрогенные артериальные гипертензии (при заболеваниях и поражениях нервной системы)
Органические поражения (сосудистые заболевания и опухоли мозга, воспалительные заболевания ЦНС (энцефалит, менингит, полиомиелит, диэнцефальный синдром), травмы мозга (посткоммоционный и постконтузионный синдром), полиневриты) нервных структур, регулирующих системную гемодинамику (нервы, продолговатый, спинной мозг, гипоталамус, кора больших полушарий) преобладание прессорных влияний над депрессорными.
Гемодинамические артериальные гипертензии (при поражении сердца и крупных сосудов)
Атеросклероз аорты поражение депрессорных зон аорты АД.
Стенозирующее поражение сонных и вертебробазилярных артерий
Коарктация аорты по закону Анрепа (сила сердечных сокращений пропорциональна давлению (сопротивлению), против которого она работает) рост УО АД.
Недостаточность аортальных клапанов рост УО по закону Франка-Старлинга (чем больше наполняется сердце во время диастолы, тем сильнее оно сокращается в систолу, т.е. ударный объем возрастает по мере увеличения наполнения сердца) АД.
Полная атриовентрикулярная блокада.
Реологическая гипертензия: увеличение вязкости крови (болезнь Вакеза, полицитэмия, эритроцитозы, гиперпротеинэмия) АД.
Васкулиты (аорто-артериит или болезнь Такаясу, васкулиты сосудов среднего и мелкого диаметра) уменьшение диаметра сосудов АД.
Особые формы вторичных артериальных гипертензий
Солевая и пищевая гипертензия (при чрезмерном употреблении соли, при употреблении веществ, богатых тирамином - некоторые сорта сыра и марки красного вина).
Медикаментозные гипертензии (при приеме глюкокортикоидов и минералокортикоидов, контрацептивных препаратов, содержащих прогестерон и эстроген, инфекундина, производных глицирризиновой кислоты (карбеноксолон), симпатических аминов, лакричного порошка, индометацина и др.).
68. – Артериальные гипотонии. Виды. Этиология. Патогенез.
Артериальная гипотония характеризуется снижением артериального давления ниже 100/60 мм рт.ст. (для лиц в возрасте до 25 лет) и ниже 105/65 мм рт.ст. (для лиц старше 30 лет).
Гипотензия артериальная ортостатическая - снижение артериального давления систолического в ортостатическом положении на 20 мм рт.ст. и более.
Физиологическая артериальная гипотония, обусловленная в основном конституциональными и наследственными факторами, встречается нередко у совершенно здоровых людей, выполняющих обычную физическую работу, и не сопровождается какими-либо жалобами и патологическими изменениями в организме. Известна физиологическая гипотония проходящего характера у спортсменов.
Патологическая артериальная гипотония
Гипотоническая болезнь (первичная артериальная гипотония, нейроциркуляторная дистония по гипотоническому типу, нейроциркуляторная гипотония) - заболевание, характеризующееся стойким снижением АД.
В основе первичной артериальной гипотонии лежит повышение тонуса парасимпатического отдела вегетативной нервной системы, нарушение функции высших вегетативных центров вазомоторной регуляции, ведущее к стойкому уменьшению общего периферического сопротивления току крови. Компенсаторное увеличение сердечного выброса в этих случаях оказывается недостаточным для нормализации артериального давления. Скорость кровотока при первичной хронической артериальной гипотонии обычно не изменена. Объём циркулирующей крови находится в пределах нормы или несколько снижен, иногда имеется склонность к нормоволемической полицитемии.
Указанные сдвиги обусловлены, скорее всего, уменьшением глюкокортикоидной активности коры надпочечников при неизменной минералкортикоидной активности.
Основное значение в возникновении гипотонической болезни, по-видимому, принадлежит длительному психоэмоциональному напряжению, в отдельных случаях – психической травме. По современным представлениям, первичная гипотония является особой формой невроза высших сосудодвигательных центров с нарушением регуляции сосудистого тонуса.
Вторичная (симптоматическая) артериальная гипотония
Выделяют острые и хронические формы.
Острые вторичные артериальные гипотензии рассмотрены в разделе «Экстремальные состояния» (шок, коллапс, синкопе).
Причины хронической вторичной артериальной гипотензии:
Значительно выраженное варикозное расширение вен нижних конечностей.
Беременность поздних сроков.
Массивный диурез.
Хроническая надпочечниковая недостаточность.
Длительный постельный режим.
Синдром гипербрадикинизма (отсутствуют киназы, расщепляющие брадикинин), наследственно обусловленный и приобретенный (при демпинг синдроме). Ортостатическая гипотензня при гипербрадикинизме возникает после еды, способствующей освобождению кининов из стенки кишечника и поджелудочной железы, и сопровождается ярким покраснением лица вследствие действия кининов на сосуды кожи.
Нарушение дуги барорефлекса на различных уровнях (при сухотке спинного мозга, B12-дефицитной анемии, хроническом алкоголизме, сахарном диабете, сирингомиелии, миелите, порфирии, полинейропатии Гийена-Барре).
Прием ганглиоблокаторов, изобарина, лабеталола, прозозина, нитратов.
69. – Патология сосудистого тонуса. Роль нервных, гуморальных, метаболических, миогенных факторов. Понятие дисфункций эндотелия и ее роль в механизмах нарушения сосудистого тонуса.
Сосудистая компонента – определяется сосудистым тонусом; существенно изменять свой диаметр способны только резистивные сосуды; интегральными показателями сосудистого тонуса являются общее периферическое сопротивление (ОПСС) и удельное периферическое сопротивление (УПСС).
Регуляция ад и сосудистого тонуса а. Местные (периферические) механизмы
Местные механизмы саморегуляции сосудистого тонуса обеспечивают адекватный кровоток в органах в зависимости от уровня метаболизма в них.
Миогенная регуляция (эффект Бейлиса).
Резкое повышение АД сопровождается сокращением гладкой мускулатуры артериол жизненно важных органов, в результате объемная скорость кровотока в этих органах не изменяется или возрастает незначительно, а при падении АД гладкие мышцы сосудов расслабляются, что позволяет поддерживать должную объемную скорость регионального кровотока.
Эндотелиальные факторы.
Сосудистый эндотелий является местом образования целого ряда соединений, участвующих в регуляции сосудистого тонуса, функции тромбоцитов и свертывания крови. В настоящее время выделяют несколько вазодилатирующих и вазоконстрикторных субстанций.
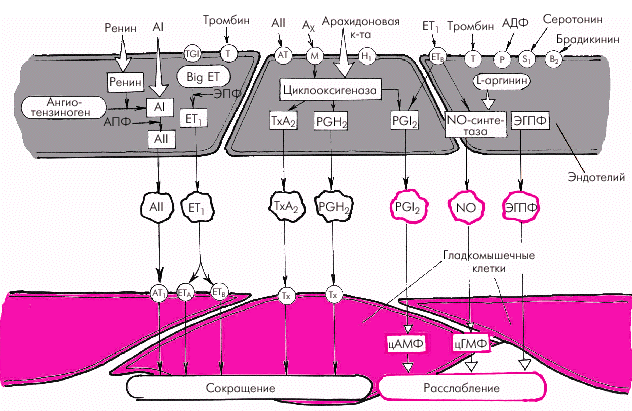
Рис. Функция эндотелия.
AI и AII - ангиотензины I и II; AX - ацетилхолин; АПФ - ангиотензин-превращающий фермент; AT1 - рецепторы к ангиотензину; B2 - рецепторы к брадикинину; Big ET - «большой» эндотелин; ET1 - эндотелин I; ETA и ETB - рецепторы к эндотелину; М - мускариновые рецепторы; H 1 - рецепторы к гистамину; NO - оксид азота; P - АДФ-рецепторы; PGI2 - простациклин; PGH 2 - простагландин H2; TxA2 - тромбоксан A2; Tx - рецепторы к тромбоксану A2; ЭГПФ - эндотелиальный гиперполяризующий фактор; ЭПФ - эндотелин-превращающий фермент.
Вазодилатирующие факторы
1. Эндотелиальный расслабляющий фактор (оксид азота, NO), обеспечивает выраженную релаксацию гладких мышц артерий, артериол и вен, а также препятствует адгезии и агрегации тромбоцитов.
Оксид азота продуцируется сосудистым эндотелием из L-аргинина спонтанно, а также при стимулировании эндотелиальной клетки рядом биологически активных веществ и медиаторов (ацетилхолин, гистамин, брадикинин, субстанция Р и др.), продукция которых возрастает в том числе при физической нагрузке и увеличении работы сердца. Освободившийся из L-аргинина оксид азота (NO) активирует гуанилатциклазу гладкомышечной клетки с образованием цГМФ, что приводит к ее активному расслаблению.
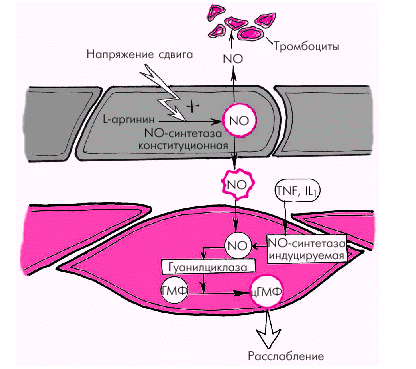
Рис. Синтез оксида азота в эндотелии. Обозначения те же. TNF – фактор некроза опухолей; IL1 - интерлейкин I.
2. Простациклин также относится к числу важнейших вазодилататоров, препятствующих вазоконстрикторному эффекту тромбоксана А2 и агрегации тромбоцитов.
Простациклин PGI2 является продуктом метаболизма арахидоновой кислоты, освобождающейся стимулированными тромбоцитами, из которой под действием циклооксигеназы синтезируется либо простациклин PGI2, либо тромбоксан А2.
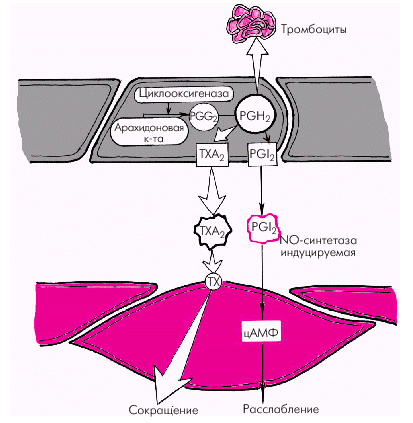
Рис. Синтез тромбоксана А2 и простациклина.
3. Эндотелиальный гиперполяризующий фактор (ЭГПФ), также вырабатываемый эндотелиальными клетками, вызывает гиперполяризацию мембран гладкомышечных клеток и снижает их чувствительность к разнообразным констрикторным влияниям.
Выделение ЭГПФ вызывает открытие калиевых каналов гладкомышечных клеток, что сопровождается расслаблением сосудов. Характерно, что в отличие от оксида азота, ЭГПФ выделяется эндотелием не постоянно, а только под действием некоторых стимулов (ацетилхолин, брадикинин, тромбин, гистамин, субстанция Р, АДФ, АТФ и др.).
Вазоконстрикторные факторы
1. Эндотелин-1 (ЕТ1) является наиболее мощным из всех известных вазоконстрикторов.
Процесс образования ЕТ1 включает несколько стадий. Вначале из предшественника эндотелинов (препроэндотелина) образуется так называемый «большой эндотелин» (проэндотелин), который, в свою очередь, под действием эндотелин-превращающего фермента (ЭПФ) трансформируется в активный полипептид, состоящий из 21 аминокислоты, - эндотелин-1 (ЕТ1). Связываясь со специфическими рецепторами клеточных мембран (ЕТА и ЕТВ), эндотелин-1 повышает внутриклеточную концентрацию ионов Са2+, что ведет к усилению сокращения гладких мышц сосудистой стенки. В физиологических условиях концентрация ЕТ1 в плазме очень мала, что связано, прежде всего, с ингибированием синтеза эндотелина-1 описанными выше вазодилатирующими субстанциями (оксидом азота и простациклином РGI2). Малые количества ЕТ1 активируют образование эндотелиальными клетками этих факторов расслабления. В более высоких концентрациях ЕТ1 стимулирует рецепторы ЕТА и ЕТВ гладкомышечных клеток, вызывая стойкую и выраженную вазоконстрикцию. Образование ЕТ1 усиливается при воздействии на эндотелиальные клетки тромбина, вазопрессина, интерлейкина-1, ангиотензина II и других веществ, а также при возникновении гипоксии, повышении АД, ускорении кровотока и т.п.
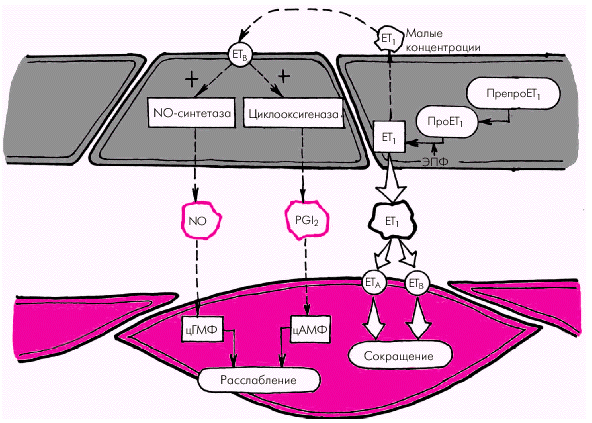
Рис. Синтез эндотелина. Обозначения те же.
2. Тромбоксан А2 и простагландин РGН2 относятся к числу активных эндотелиальных вазоконстрикторов, обладающих также свойством активировать агрегацию тромбоцитов и тромбообразование.
Являются продуктами метаболизма арахидоновой кислоты, они присутствуют во многих тканях организма, в том числе в сосудистом эндотелии.
3. Тканевой ангиотензин II (АII) относится к числу мощных вазоконстрикторов, образующихся в эндотелии различных сосудистых областей.
Он образуется из неактивного ангиотензина I (АI) под действием тканевого ангиотензин-превращающего фермента (АПФ). Этот фермент присутствует в эндотелиальных клетках, что обеспечивает образование АII непосредственно на поверхности эндотелия. Взаимодействуя со специфическими ангиотензиновыми рецепторами (АТ1) гладкомышечных клеток, он также увеличивает внутриклеточную концентрацию Са2+, усиливая сокращение гладких мышц сосудистой стенки.
В физиологических условиях существует оптимальное соотношение выработки эндотелиальных вазодилатирующих и вазоконстрикторных субстанций, которое полностью соответствует метаболическим потребностям органа и основным параметрам центральной гемодинамики.
При действии на сосудистый эндотелий различных повреждающих факторов (гипоксии, чрезмерной концентрации катехоламинов, ангиотензина II, серотонина, высокого уровня АД, ускорения кровотока и др.) начинают преобладать вазоконстрикторные механизмы регуляции сосудистого тонуса, и развивается так называемая дисфункция эндотелия. Она характеризуется повышением тонуса сосудистой стенки, ускорением агрегации тромбоцитов, процессов пристеночного тромбообразования и т.п.
Продукты метаболизма.
В интенсивно работающем органе под действием продуктов метаболизма (ионов Н+, аденозина, АТФ, АДФ, АМФ, СО2, молочной кислоты и др.) и биологически активных веществ (брадикинина, гистамина и др.) также происходит снижение тонуса артериол и прекапиллярных сфинктеров и увеличивается, таким образом, число функционирующих капилляров. Наоборот, при снижении метаболизма эти эффекты уменьшаются, и происходит адекватное ограничение органного кровотока.
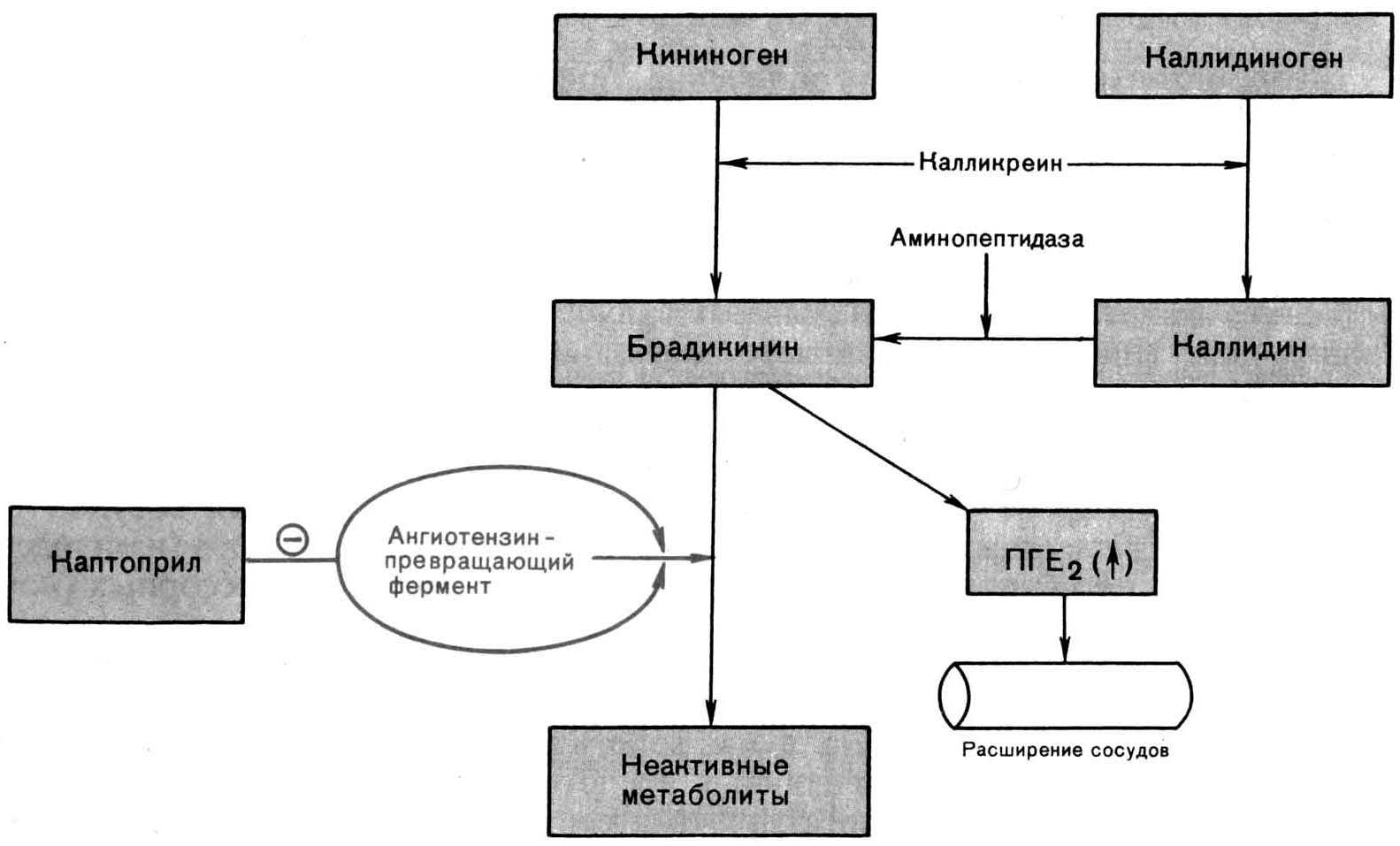
Рис. Влияние каптоприла на метаболизм брадикинина. (+) - повышение концентрации; (-) - угнетающее действие; ПГЕ2 - простагландин Е2.
Б. Центральные механизмы регуляции
Афферентное звено.
Представлено многочисленными баро- и хеморецепторами, расположенными в нескольких рефлексогенных зонах сосудистой системы (аорта, синокаротидная зона, сосуды легких и др.).
Рис. Баро- и хеморецепторы аорты и каротидного синуса.
Барорецепторы реагируют на степень и скорость растяжения стенки сосудов (или полостей сердца). При повышении АД или наполнения камер сердца барорецепторы отвечают усилением афферентной импульсации, при снижении АД - ее уменьшением.
Хеморецепторы дуги аорты, синокаротидной зоны и других рефлексогенных зон (сердце, почки, органы пищеварения) аналогично реагируют на изменение в крови концентрации О2, СО2, ионов Н+.
Чувствительные волокна от баро- и хеморецепторов дуги аорты и каротидного синуса проходят в составе синокаротидного нерва, ветвей языкоглоточного нерва и депрессорного нерва.
Центральное звено.
Центральное звено регуляции сосудистого тонуса – вазомоторный (сосудо-двигательный) центр - представлено различными функционально связанными между собой нервными структурами, расположенными в продолговатом, спинном мозге, гипоталамусе, коре больших полушарий.
Известна так называемая ишемическая реакция ЦНС. При значительном снижении системного АД (около 40 мм рт.ст.) возникает ишемия сосудо-двигательного центра и активация симпатической нервной системы. Медиатором последней является норадреналин, вызывающий тахикардию (1-рецепторы) и увеличение тонуса сосудов(1 и 2-рецепторы).
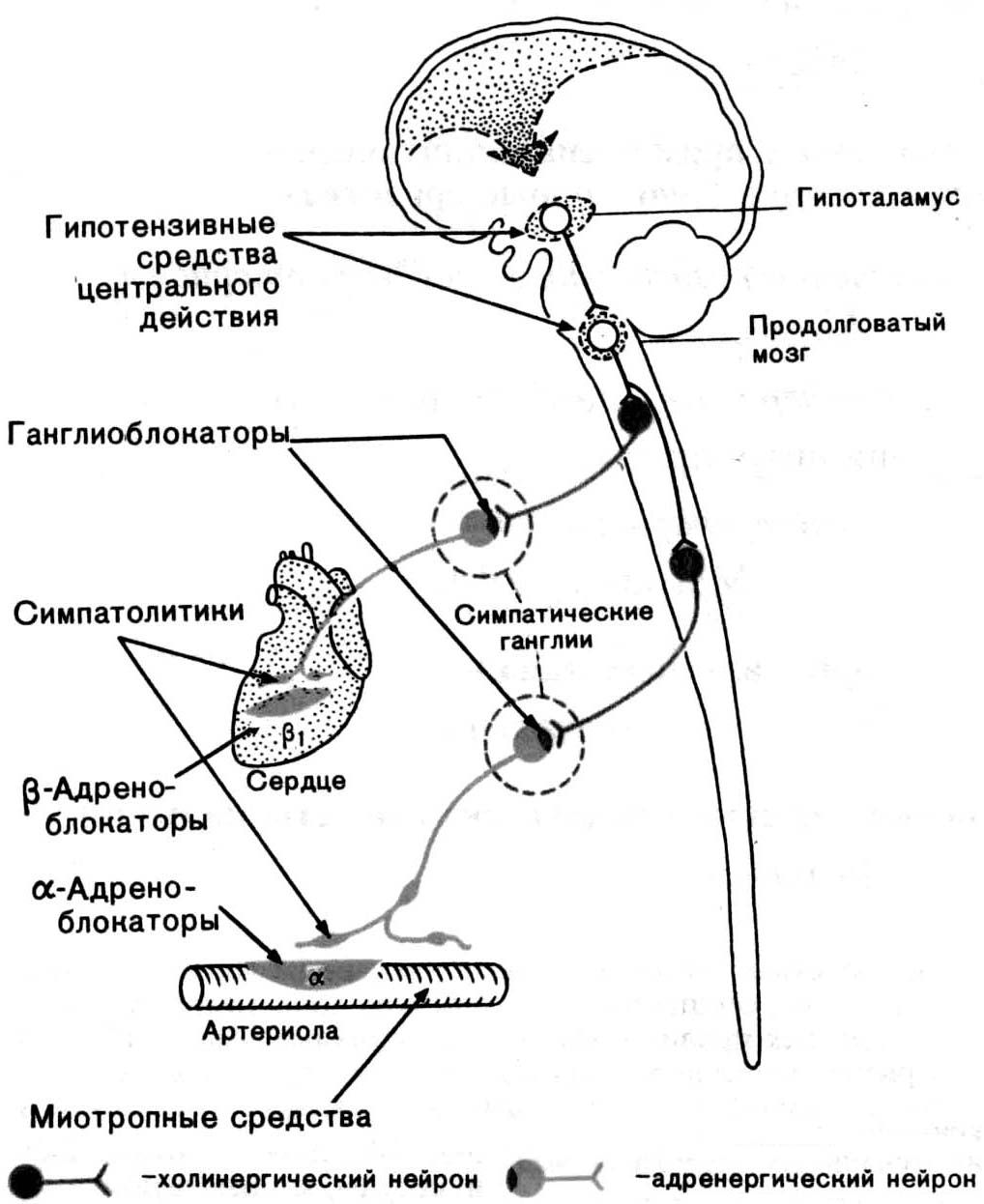
Рис. Схема симпатической иннервации сосудов и сердца с указанием локализации действия нейротропных и миотропных гипотензивных средств.
Эфферентное звено.
Включает нервные и гуморальные механизмы регуляции сосудистого тонуса. В зависимости от скорости развития циркуляторных эффектов различают: 1) механизмы быстрого кратковременного действия; 2) механизмы промежуточного действия; 3) механизмы длительного действия.
К механизмам быстрого кратковременного действия относятся нервные рефелекторные реакции, возникающие при раздражении баро- и хеморецепторов описанных рефлексогенных зон, а также при ишемии ЦНС. Эти реакции развиваются в течение нескольких секунд и реализуются через рефлекторные изменения активности симпатической и парасимпатической нервных систем, а также через изменение концентрации гуморальных веществ - адреналина и норадреналина. Раздражение барорецепторов аорты и каротидного синуса (например, при повышении АД или механическом воздействии на эти зоны) закономерно приводит к снижению симпатических (вазоконстрикторных) и усилению парасимпатических (депрессорных) влияний. В результате снижается сосудистый тонус, а также частота и сила сокращения сердца, что способствует нормализации АД. Наоборот, при падении АД (например, при кровопотере) импульсация с барорецепторов уменьшается, и начинают преобладать симпатические влияния - увеличение ЧСС, сердечного выброса и сосудистого тонуса.
Аналогичным образом возникает ответ на раздражение рецепторов растяжения предсердий и рецепторов растяжения желудочков, например, при быстром увеличении их наполнения. В результате снижения тонуса симпатических и повышения активности парасимпатических нервов развивается брадикардия и вазодилатация.
Возбуждение хеморецепторов дуги аорты и каротидного синуса при снижении напряжения О2, повышении напряжения СО2 или увеличении концентрации ионов Н+ в крови приводит к сужению резистивных сосудов и подъему АД. К такому же эффекту приводит рефлекторная реакция на ишемию ЦНС, например, при недостаточном кровоснабжении головного мозга, гипоксемии или резком падении АД. Повышение концентрации Н+ и СО2 сопровождается раздражением хеморецепторов ствола мозга и значительным подъемом АД.
Симпатической нервной системе принадлежит ведущая роль в регуляции тонуса периферических сосудов. Влияние адреналина и норадреналина на тонус различных сосудистых областей зависит от концентрации этих веществ в крови и от соотношения в разных сосудах - и -адренорецепторов. Как известно, возбуждение -рецепторов сопровождается сокращением гладких мышц, а возбуждение -рецепторов - их расслаблением.
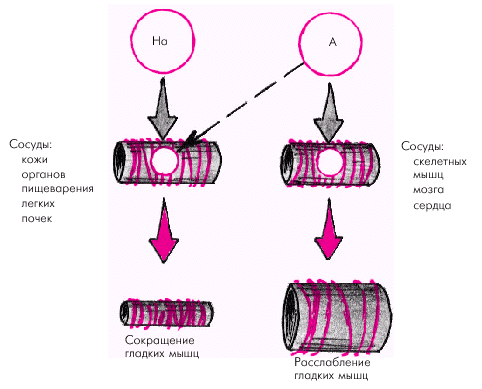
Рис. Влияние норадреналина (На) и адреналина (А) на - и -рецепторы сосудов и различные циркуляторные эффекты.
Норадреналин воздействует преимущественно на -адренорецепторы, вызывая в экстремальных условиях увеличение сосудистого тонуса, системного периферического сопротивления и АД. Адреналин взаимодействует как с -, так и с -адренорецепторами. В физиологических концентрациях он возбуждает преимущественно -рецепторы, вызывая расслабление гладкой мускулатуры сосудов, особенно тех из них, в которых преобладают -адренорецепторы (скелетные мышцы, мозг, сердце). Одновременно адреналин повышает УО и ЧСС, в результате чего в обычных физиологических условиях уровень системного АД под действием адреналина существенно не меняется.
В экстремальных ситуациях (сильный эмоциональный стресс, острое кровотечение и т.п.), когда концентрация адреналина в крови повышается в десятки раз, может проявляться его взаимодействие с -адренорецепторами сосудов и преобладать сосудосуживающие реакции (особенно в коже, органах пищеварения и легких, в которых имеется большое количество -рецепторов).
Главным регуляторным механизмом промежуточного действия является почечная ренин-ангиотензиновая система (РАС). Ее активация, наступающая при снижении кровоснабжения почек любого генеза (падение АД, сужение почечных сосудов и т.п.), сопровождается выделением ренина, который способствует превращению ангиотензиногена в ангиотензин I. Последний под действием АПФ превращается в ангиотензин II, обладающий мощным вазоконстрикторным действием. Кроме того, ангиотензин II возбуждает центральные и периферические симпатические структуры. Все это приводит к росту периферического сопротивления и повышению (нормализации) АД. Следует помнить, что существует альтернативный путь трансформации АI в АII, без участия АПФ.
Рис. Схема активации ренин-ангиотензиновой системы при снижении артериального давления. АПФ - ангиотензин-превращающий фермент.
К регуляторным механизмам длительного действия относят почечные системы контроля за объемом жидкости, гормональные системы альдостерона, вазопрессина, тироксина, глюкокортикоидов.
Компенсаторные механизмы при изменении АД
Бароцептивный рефлекс: при АД в дуге аорты и каротидном синусе через активацию блуждающего нерва ЧСС.
Рефлекс Берцольда-Яриша: АД раздражение рецепторов эндокарда через активацию блуждающего нерва ЧСС.
Возбуждение тормозных 2-адренорецепторов на пресинаптической мембране 1-адренергических синапсов сосудов пресинаптическое торможение.
При АД диурез.
Адреналин ослабляет канальцевую реабсорбцию Na+ диурез.
тонуса сосудов ишемия почек выделение почечных простагландинов (медуллин) расширение сосудов.
Выделение в предсердиях НУГ (Na-уретический гормон) клубочковой фильтрации.
тонуса сосудов ишемия почек выделение ренина в ЮГА … сужение сосудов + альдостерон гиперосмолярность плазмы вазопрессин еще большее АД.
В клинической практике наиболее часто используются следующие виды АД крови: минимальное (диастолическое), пульсовое, среднее гемодинамическое и максимальное (систолическое) давление.
Рис. Давление крови в аорте.
Диастолическое давление представляет собой величину минимального давления крови, достигаемую к концу диастолического периода сердечного цикла. Минимальное давление зависит от степени проходимости или величины оттока крови через систему прекапиляров, упруго-вязких свойств артериальных сосудов.
Систолическое давление равно максимальному давлению, достигаемому в момент, соответствующий выбросу крови из сердца в аорту. Максимальное давление характеризует запас энергии, которым обладает движущаяся масса крови на данном участке сосуда.
Среднее гемодинамическое давление определяется интегрированием (усреднением) текущего значения АД за время сердечного цикла.
Ориентировочно величину среднего давления можно определить по формулам:
Вецлера и Богера: Рm = 0,42 Рs + 0,58 Рd,
Хикема: Pm = Pd + (Ps - Pd) / 3,
где Рs - систолическое (максимальное) давление, Рd - диастолическое (минимальное) давление.
70. – Этиология дыхательной недостаточности. Нарушения альвеолярной вентиляции, перфузия легких, вентиляционно-перфузионных отноше5ний, диффузии газов в легких.
Дыхательная недостаточность - патологическое состояние, развивающееся вследствие нарушения внешнего дыхания, при котором не обеспечивается поддержание адекватного условиям газового состава артериальной крови.
Острая
Умеренная (PaO2 > 70 мм рт.ст.).
Средняя (PaO2 = 50-70 мм рт.ст.).
Тяжелая (PaO2 < 50 мм рт.ст.).
Хроническая
Компенсированная (HbO2N 95%).
Субкомпенсированная (HbO2 < 93%).
Декомпенсированная (HbO2 < 75%).
Патофизиологическая классификация нарушений внешнего дыхания
Нарушения альвеолярной вентиляции:
альвеолярная гиповентиляция
нарушение биомеханики дыхания (по обструктивному или рестриктивному типу),
нарушение регуляции дыхания,
альвеолярная гипервентиляция
активная,
пассивная.
Нарушения перфузии легких:
легочная гипертензия,
легочная гипотензия.
Нарушение вентиляционно-перфузионных отношений.
Нарушение диффузионной способности легких.
Смешанные нарушения.
Асфиксия (в переводе с греч. – «без пульса») - состояние гипоксии, сочетающееся с повышением напряжения углекислого газа в крови и тканях. Сопровождается тяжелыми расстройствами нервной, дыхательной, сердечно-сосудистой систем (обусловлены недостатком О2 и избытком СО2).
Механическая асфиксия (при препятствии наступлению воздуха в дыхательные пути):
Воспалительные процессы (отек гортани, дифтерия)
Опухоли
Спазмы
Западение языка
Аспирация воды, рвотных масс, пищи, крови
Попадание инородных тел
Сдавление извне - травматическая асфиксия
Асфиксия в замкнутых пространствах
Стадии острой механической асфиксии
Усиленная деятельность дыхательного центра (инспираторная одышка): повышение АД; ЧСС; возбуждение симпатической нервной системы; расширены зрачки; демонохронизация на ЭЭГ.
Экспираторная одышка: АД повышается; ЧСС замедляется (медленные высокоамплитудные сокращения - вагус-пульс). Преобладание парасимпатической нервной системы, зрачки сужены, угнетение биотоков мозга.
В 1-ю и 2-ю стадии преобладает возбудительный процесс.
Тормозной процесс: претерминальная остановка дыхания, АД начинает падать, угасают рефлексы, исчезают биотоки мозга.
Терминальное дыхание (гаспинг-дыхание): судороги, непроизвольное мочеиспускание и дефекация, понижение АД и ВД (венозное давление), смерть от паралича дыхания, сердцебиение продолжается еще 5-8 мин.
Иногда 1-я стадия отсутствует (при повешении, утоплении). Может быть остановка сердца и дыхания при раздражении рецепторных систем (при удавлении)
У перенесенных асфиксию развиваются пневмонии, отек легких, парез голосовых связок, ретроградная амнезия.
Дефицит возбуждающей афферентации.
Синдром асфиксии новорожденных - незрелость хеморецепторов у недоношенных детей (для дополнительной стимуляции дыхательного центра используют похлопывание по ягодицам, обрызгивание холодной водой).
Синдром Пиквика – сонливость с гиповентиляцией и апное (снижение тонуса ретикулярной формации и повышение порога возбудимости центральных хеморецепторов).
Избыток тормозной афферентации.
Остановка дыхания на вдохе при раздражении слизистой верхних дыхательных путей, например, при остром респираторном заболевании, действии химических и механических агентов (срабатывает тормозной тригемино-вагусный рефлекс Кречмера).
Сильные болевые ощущения, сопровождающие акт дыхания (травма грудной клетки, плеврит, воспаление дыхательных мышц).
Избыток возбуждающей афферентации.
«Перевозбуждение» ДЦ может характеризоваться развитием альвеолярной гиповентиляция при тахипноэ, что является следствием увеличения функционального мертвого пространства.
Стрессорные воздействия.
Неврозы (например, приступы истерии).
Некоторые поражения структур среднего мозга.
Нарушения кровообращения.
Острое воспаление.
Механическая травма и др.
Избыток афферентации рефлекторного происхождения иногда возникает при раздражении брюшины, термических или болевых воздействиях на кожные покровы.
Хаотическая афферентация.
Поступление к ДЦ и мотонейронам дыхательных мышц различных афферентных влияний, имеющих хаотический (неупорядоченный) характер по критерию обеспечения газообменной функции легких, что возможно во время пения, игры на духовых инструментах, у стеклодувов и т.д.
Формирование мощных потоков афферентной импульсации различной модальности (болевой, психогенной, хеморецепторной, барорецепторной и пр.), нарушающие нормальный дыхательный ритмогенез (при обширных травмах, ожогах, в остром периоде инфаркта миокарда, висцеральных повреждениях).
Патология дыхательного центра
Энцефалиты.
Нарушения мозгового кровообращения.
Опухоли мозга.
Травмы мозга.
Отек мозга.
Интоксикации мозга.
Патология дыхательного центра проявляется патологическими типами дыхания (см. выше).
Патология эфферентных путей
Травмы шейного отдела спинного мозга - сохраняется естественное диафрагмальное дыхание, но нарушается иннервация дыхательных межреберных мышц.
Повреждение диафрагмальных мотонейронов (при сирингомиелии, рассеянном склерозе, полиомиелите) - сохраняется только произвольное дыхание; формируется синдром "проклятия Ундины" (отсцтствие непроизвольного дыхания остановка дыхания при засыпании).
Повреждение в области N-холинорецепторов: ятрогенное (при передозировке миорелаксантов), при миастении (слабость дыхательных мышц из-за врожденного или аутоиммунного повреждения N-холинорецепторов).
Миогенные расстройства
Дистрофия Дюшена.
Миозиты.
Паралич диафрагмы.
Альвеолярная гипервентиляция
Пассивная гипервентиляция обусловлена неправильным подбором режима искусственной вентиляции.
Активная гипервентиляция обусловлена избытком афферентации:
психогенная (истерия),
церебральная (опухоли и т.д.),
рефлексогенная (травмы).
Нарушения перфузии легких, вентиляционно-перфузионных отношений, диффузионной способности легких.
Нарушение перфузии легких
Легочная гипертензия
Прекапиллярная форма
Тромбоэмболия легочной артерии (ТЭЛА), см. тему «Нарушения регионального кровообращения. Тромбозы и эмболии».
Рефлекс Эйлера (снижение pO2 в альвеолах ведет к гипоксической вазоконстрикции), см. выше.
Посткапиллярная форма (застой крови) при левожелудочковой недостаточности (митральный стеноз, кардиосклероз, ГБ, ИМ)
Смешанная форма
Рефлекс Китаева при сердечной недостаточности ( давления в левом предсердии спазм легочных артериол).
ДМПП и ДМЖП со сбросом слева направо.
Легочная гипотензия (коллапс, шок, пороки Фалло), см. темы «Экстремальные состояния», «Сердечная недостаточность).
Нарушение вентиляционно-перфузионных отношений
Соотношение между вентиляцией и кровотоком принято характеризовать с помощью так называемого показателя вентиляционно-перфузионных отношений (VА/QТ), который в норме равен 0,8-1,0, что отражает адекватность минутного объема альвеолярной вентиляции минутному объему кровотока в легких.
Сосудистая патология. Шунтирование.
Снижение показателя вентиляционно-перфузионных отношений наблюдается при локальной альвеолярной гиповентиляции:
расстройства обструктивного типа,
нарушения эластичности легочной ткани,
локальное неравномерное действие сил вдоха и выдоха,
парадоксальное или маятникообразное дыхание при одностороннем параличе диафрагмальной мышцы,
легочно-плевральных сращения,
деформация грудной клетки.
Увеличение показателя вентиляционно-перфузионных отношений наблюдается при локальной закупорке, стенозе (облитерации) или спазме сосудов системы легочной артерии.
Нарушение диффузионной способности легких Диффузия газов
|
О2, мм рт.ст |
СО2, мм рт.ст |
Атмосферный воздух |
150 |
0,2 |
Альвеолярный воздух |
100 |
40 |
Выдыхаемый воздух |
114 |
29 |
Артериальная кровь |
100 |
40 |
Венозная кровь |
40 |
46 |
Закон диффузии Фика - диффузионный поток прямо пропорционален градиенту концентрации веществ.
Коэффициент диффузии для СО2 в 23 раза больше, чем для О2.
Диффузионная способность легких (D), в норме равна 30 мл/мин.мм рт.ст.
VO2
D = , где
PO2
Vo2 - количество поглощенного кислорода, Pо2 - средний градиент парциального давления O2 между альвеолярным пространством и кровью легочных капилляров.
Эффективность газообмена в альвеолах
D/Q, где
Q - показатель легочной перегрузки.
Причины нарушения диффузии:
Диффузный фиброзирующий альвеолит (болезнь Хаммана-Рича).
Недостаток сурфактанта («синдром гиалиновых мембран» у новорожденных).
Пневмокониоз (силикоз, асбестоз, бериллиоз) - фиброз легких в результате длительного вдыхания пыли.
Воспалительные и токсические поражения.
71. – Этиология и патогенез обструктивных и реструктивных нарушений вентиляции легких. Принципы функциональной диагностики.
Этиология и патогенез обструктивных и рестриктивных типов нарушения вентиляции легких. Легочное сердце (понятие).
Для осуществления эффективной легочной вентиляции необходимо два условия:
беспрепятственное прохождение воздуха по бронхиальному дереву до респираторного отдела легких;
наличие достаточного количества альвеол, способных к газообмену, и адекватное увеличение их объема при дыхании, т.е. наличие достаточной площади альвеолярно-капиллярной мембраны, через которую происходит газообмен.
Соответственно этим положениям выделяют два типа нарушения вентиляции легких.
Обструктивный тип связан с нарушением прохождения воздуха по бронхиальному дереву (повышение аэродинамического сопротивления в бронхах), обусловленным:
спазмом гладкой мускулатуры бронхов - бронхоспазм (бронхиальная астма),
воспалительной инфильтрацией и отеком слизистой бронхов (бронхит),
гиперсекрецией слизи,
врожденной и приобретенной деформацией бронхов (бронхоэктатическая болезнь),
обтурацией бронхов (кровь, экссудат, опухоли, инородные тела бронхов),
экспираторным коллапсом мелких бронхов (компрессия мелких бронхов на выдохе при затрудненном выдохе) при бронхитах, бронхиальной астме, эмфиземе легких.
Рестриктивный (ограничительный) тип - связан либо с уменьшением суммарной площади альвеолярно-капиллярного газообмена, либо со снижением способности легочной ткани к растяжению при дыхании (чаще всего эти две причины взаимосвязаны):
Собственно заболевания органов дыхания:
инфильтративные изменения легочной ткани,
пневмосклероз,
уменьшение объема функционирующей паренхимы легкого (резекция легкого, ателектаз, врожденная гипоплазия легкого, заболевания плевры, ограничивающие экскурсию легкого).
Внелегочные нарушения:
деформация грудной клетки и позвоночника,
нарушения деятельности дыхательной мускулатуры,
венозная гиперемия легких при левожелудочковой недостаточности,
увеличение объема брюшной полости,
болевой синдром, приводящий к ограничению подвижности диафрагмы.
В клинических условиях при заболевании органов дыхания имеется сочетание обструктивных и рестриктивных нарушений, т.е. комбинированная вентиляционная недостаточность с преобладанием одной из форм. Выделение этих форм помогает понять ведущий механизм вентиляционной недостаточности и назначить патогенетически обоснованное лечение.
Легочное сердце - гипертрофия правого желудочка при заболеваниях легких.
Синдром легочного сердца развивается при ухудшении аэрации альвеол (патология бронхов и легких, ТЭЛА, васкулиты) срабатывает рефлекс Эйлера (при нарушении вентиляции альвеол развивается спазм легочных артериол перегрузка ПЖ давлением гипертрофия ПЖ.
72. – Патофизиологическое значение нарушений желудочной секреции и моторики. Виды нарушений.
В кишечнике осуществляется дистантное (полостное) и мембранное (пристеночное) пищеварение.
Поступление HCl, жиров, белков, углеводов и частично переваренных пищевых продуктов из желудка в верхнюю часть двенадцатиперстной кишки вызывает секрецию, по крайней мере, пяти разных гормонов: секретина, холецистокинина, желудочного ингибиторного пептида, мотилина, вазоактивного интестинального пептида, энтероглюкогона, соматостатина, участвующих в регуляции процессов пищеварения.
Суть полостного пищеварения заключается в разрушении крупных молекул, поступивших из желудка, под действием ферментов поджелудочной железы: трипсиногена, химотрипсиногена, аминопептидазы, дипептидазы, проэластазы. Эти ферменты работают уже в слабо щелочной среде.
Нарушение кишечного полостного пищеварения
Ахолия – уменьшение или полное прекращение поступления желчи (и соответственно желчных кислот) в кишечник.
Причины ахолии: недостаточность образования (гепатит, цирроз), нарушение желчеотделения (закупорка камнями, сужение, сдавление, дискинезия желчевыводящих путей).
Функции желчи:
усиливает действие ферментов панкреатического сока (трипсина, амилазы), активирует липазу, принимает участие в эмульгировании жиров;
играет важную роль в процессе всасывания жирных кислот, жирорастворимых витаминов A, D, Е, К, аминокислот, холестерина, солей кальция;
повышает тонус и усиливает перистальтику кишечника;
оказывает бактериостатическое действие на кишечггую микрофлору, предупреждая развитие гнилостных процессов;
участвует в пристеночном пищеварении, создавая благоприятные условия для фиксации ферментов на мембранах микроворсинок энтероцитов.
Расстройства кишечного пищеварения при ахолии:
снижение переваривания и всасывания жиров;
нарушение расщепления белков и углеводов;
ослабление перистальтики толстой кишки;
создаются благоприятные условия для развития гнилостных и бродильных процессов в кишечнике;
снижение всасывания витамина К;
нарушение темпа эвакуации желудочного содержимого в кишечник.
Вследствие отсутствия желчных пигментов кал обесцвечивается. При ахолии развивается стеаторея.
Стеаторея – симптом, сопровождающий нарушение переваривания липидов, которые выводятся с калом.
Причины стеатореи: дефицит панкреатической липазы (панкреатическая), снижение секреции желчи (гепатогенная), угнетение ресинтеза триглицеридов в кишечнике при его заболеваниях (энтерогенная).
Панкреатическая ахилия – нарушение секреции панкреатического сока, содержащего основные пищеварительные ферменты.
Причины панкреатической ахилии: органические поражения поджелудочной железы (хронический панкреатит, опухоль), закупорка камнем или сдавление опухолью протока поджелудочной железы, нарушение нейрогуморальной панкреатической регуляции и секреции; функциональная недостаточность поджелудочной железы может возникать при некоторых эндокринопатиях, инфекционных заболеваниях
Нарушение кишечного мембранного пищеварения
В процессах мембранного пищеварения принимают участие кишечные ферменты, синтезируемые энтероцитами, а также ферменты панкреатического происхождения, адсорбированные на мембранах микроворсинок.
Особенности мембранного пищеварения:
высокая сопряженность ферментативного расщепления пищевых веществ и их всасывания (завершающий этап переваривания происходит на поверхности мембран, через которые затем осуществляется транспорт конечных продуктов гидролиза);
высокая скорость переваривания пищевых субстратов (благодаря образованию фермент-мембранных комплексов): ферменты кишечных клеток и поджелудочной железы фиксируются на клеточных мембранах ворсинок;
энтеропептидаза вырабатывается клетками слизистой и активирует трипсиноген непосредственно у стенки кишечника;
микробы просвета кишечника не могут использовать аминокислоты, сахара, жирные кислоты, т.к. размеры микробов больше просветов между выростами щеточной каемки.
Причины нарушения мембранного пищеварения
Недостаточность желчевыделения или уменьшение секреции панкреатического сока приводят в основном к нарушениям полостного пищеварения в тонкой кишке. Расстройства полостного пищеварения в свою очередь могут явиться причиной нарушения мембранного (пристеночного) пищеварения.
Поражения слизистой кишечника (энтериты, лучевые и лекарственные поражения, опухоли) структурные изменения зоны щеточной каймы, ее примембранного слоя (гликокаликса), самих ворсинок.
Изменения ферментативного слоя кишечной поверхности и нарушениях активного транспорта питательных веществ (гипофункция надпочечников, лучевая болезнь, интоксикации, снижение желчевыделения, голодание).
Врожденный дефицит кишечных гидролаз.
I группа: в основном связана с недостаточностью лактазы. Т.к. дисахарид лактоза встречается только как компонент молока, то это имеет значение в развитии расстройств у детей грудного возраста. Непереваренная лактоза недоступна организму, она остается в кишечнике, используется бактериями и поддерживает развитие патогенной кишечной флоры. Отсюда - метеоризм, понос и рвота с последующей изотонической дегидратацией и алкалозом. Этому заболеванию сопутствует потеря аминокислот с мочой, что влечет нарушение белкового обмена и гипотрофия. Единственный выход - переход на искусственное вскармливание.
Непереносимостью лактозы страдают и взрослые люди. У них прием молока также вызывает желудочно-кишечные расстройства. Недостаточность лактазы у таких людей развивается в онтогенезе, т.е. не сразу после рождения.
II группа: непереносимость сахарозы. Этиология - недостаточность сахаразы. Патогенез связан с развитием мощной флоры кишечника и энтеритом. Клиника та же, но проявляется позже, т.е. когда ребенок переходит на смешанное вскармливание и когда молочный сахар (лактоза) заменяется сахарозой. Развивается хроническая диарея, гипотрофия, в тяжелых случаях - гибель ребенка.
III группа: непереносимость мальтозы, нарушение мальабсорбции. Этиология - недостаточность мальтазы. Патогенез и клиника аналогичны первой и второй группам. Лечение - исключение из пищи соответствующего дисахарида. Диагноз дисахаридозов ставится на основании обнаружения в кале дисахаридов.
Заболевания поджелудочной железы.
Двигательные расстройства тонкой кишки и микроворсинок (повышение тонуса блуждающего нерва, снижение синтеза интерстициального гормона вилликинина).
Генерализованные нарушения всасывания
Атрофия ворсинок, т.к. уменьшается площадь всасывающей поверхности. Состояние может индуцироваться повышенной чувствительностью к клейковине (глютеновая энтеропатия), при тропическом спру, идиопатической стеаторее.
Хирургическая резекция значительной части тонкого кишечника.
Обширный инфильтрат и воспаление слизистой кишечника (как правило, на фоне измененной микрофлоры).
После гастрэктомии.
Карциноидный синдром, который связан с образованием избытка серотонина опухолями, содержащими аргентаффинные клетки, и возникающими обычно в тонком кишечнике, а также их метастазами, локализованными, как правило, в печени. Недостаточность всасывания, видимо, обусловлена стимуляцией перистальтики серотонином и быстрым пассажем пищи.
Заболевания поджелудочной железы - нарушение всасывания обусловлены недостаточностью переваривания и затрагивает преимущественно высокомолекулярные соединения.
Прием некоторых лекарственных препаратов.
Неомицин вызывает обратимую ингибицию деятельности дисахаридаз, приводит к повреждению слизистой оболочки тонкой кишки и неблагоприятно воздействует на процесс всасывания жиров, нарушая процесс образования мицелл.
Колхицин приводит к остановке процесса деления клетки в метафазе, нарушая тем самым нормальный процесс деления клеток слизистой оболочки тонкой кишки.
Холестирамин и колестипол связываются с желчными кислотами, появляется повышенный риск развития нарушения всасывания жиров. У пациентов, которые длительное время принимают холестирамин (без поддерживающего лечения витамином D и препаратами кальция), развивается нарушение всасывания жирорастворимых витаминов, что сопровождается развитием остеомаляции.
Прием больших количеств антацидных препаратов, содержащих алюминий, вызывает нарушение всасывания фосфора, что, в свою очередь, вызывает тяжелые метаболические поражения костной ткани (развитие остеомаляции) и нарушение функции почек.
Хронический прием слабительных препаратов в избыточных количествах также может приводить к возникновению различных нарушений всасывательной функции кишки и синдрому мальабсорбции.
Нарушения двигательной функции кишечника
Усиление перистальтики кишечника происходит
при воспалении (энтерит, колит);
под влиянием механического, либо химического раздражения плохо переваренной пищей;
под действием бактериальных токсинов;
при расстройстве нервной и гуморальной регуляции;
при повышении тонуса блуждающего нерва;
усилении секреции серотонина, гастрина, мотилина;
снижении секреции вазоактивного интестинального пептида.
Клиническим выражением этих воздействий является диарея, которая в ряде случаев может играть защитную роль. Однако длительный и обильный понос может закончиться эксикозом и даже коллапсом.
Ослабление перистальтики кишечника наступает
при малом объеме пищи с недостаточным содержанием клетчатки;
при повышенном переваривании ее в верхних отделах ЖКТ;
при пониженной возбудимости центра блуждающего нерва.
снижении секреции серотонина, гастрина, мотилина;
усилении секреции вазоактивного интестинального пептида.
В итоге развиваются запор, застой кала, брожение, гниение, метеоризм.
Основная функция толстого кишечника - реабсорбция электролитов и воды. Главным фактором изменения количества жидкости и ионов в содержимом толстого кишечника является, по-видимому, реабсорбция натрия за счет его активного транспорта. При колитах, т.е. воспалительных поражениях толстого кишечника, нарушаются многие обменные процессы, не говоря уже о малоприятных симптомах этого заболевания.
Рвота – непроизвольный выброс содержимого пищеварительного тракта через рот (иногда и нос).
Рвота начинается сокращениями тонкой кишки, в результате чего часть ее содержимого антиперистальтическими волнами выталкивается в желудок. Через 10-20 с происходят сокращения желудка, раскрывается кардиальный сфинктер, после глубокого вдоха сильно сокращаются мышцы брюшной стенки и диафрагмы, вследствие чего содержимое в момент выдоха выбрасывается через пищевод в полость рта; рот широко раскрывается, и из него удаляются рвотные массы. Их попадание в воздухоносные пути обычно предотвращено остановкой дыхания, изменением положения надгортанника, гортани и мягкого неба.
Центр рвоты расположен на дне IV желудочка в ретикулярной формации продолговатого мозга. Он связан с центрами других отделов мозга и центрами других рефлексов.
Импульсы к центру рвоты поступают от многих рецепторных зон.
корня языка, глотки,
слизистой оболочки желудка, желчных путей,
брюшины,
коронарных сосудов,
вестибулярного аппарата (при укачивании),
мозга.
Рвота может быть обусловлена действием обонятельных, зрительных и вкусовых раздражителей, вызывающих чувство отвращения (условнорефлекторная рвота).
Ее также вызывают некоторые вещества, действующие гуморально на нервный центр рвоты. Эти вещества могут быть эндогенными и экзогенными.
Эфферентные импульсы следуют к кишечнику, желудку и пищеводу в составе блуждающего и чревного нервов, а также нервов, иннервирующих брюшные и диафрагмальные мышцы, мышцы туловища и конечностей, что обеспечивает основные и вспомогательные движения (в том числе и характерную позу).
Рвота сопровождается изменением дыхания, кашлем, потоотделением, слюноотделением и другими реакциями.
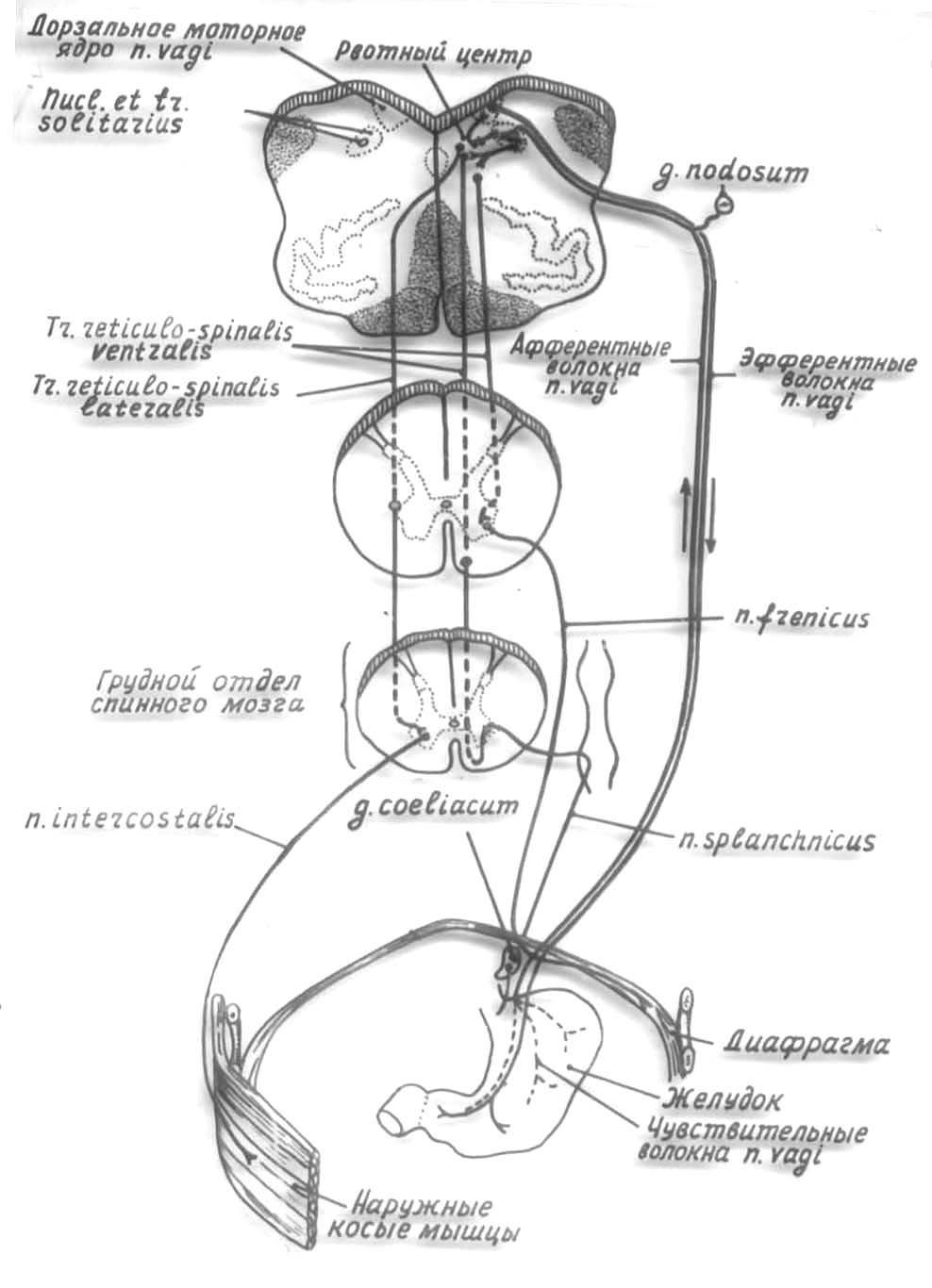
73. – Современные представления о этиологии язвенной болезни желудка и двенадцатиперстной кишки. Патогенез демпинг синдромов.
С современных позиций язвенная болезнь (ЯБ) рассматривается как полиэтиологическое, генетически и патогенетически неоднородное заболевание.
Рис. Хроническая язва желудка.
К настоящему времени доказаны три основные причины язвенной болезни:
Местная инфекция бактериями Helicobacter pylori (H. pylori). Заболевание начинается с попадания бактерии в желудок через рот. Там она внедряется в защитный слой слизи и прикрепляется к апикальной части клеток эпителия. Наиболее активно заселяется бактериями антральный отдел желудка и луковицы двенадцатиперстной кишки.
Рис. Бактерия Helicobacter pylori на стенке двенадцатиперстной кишки.
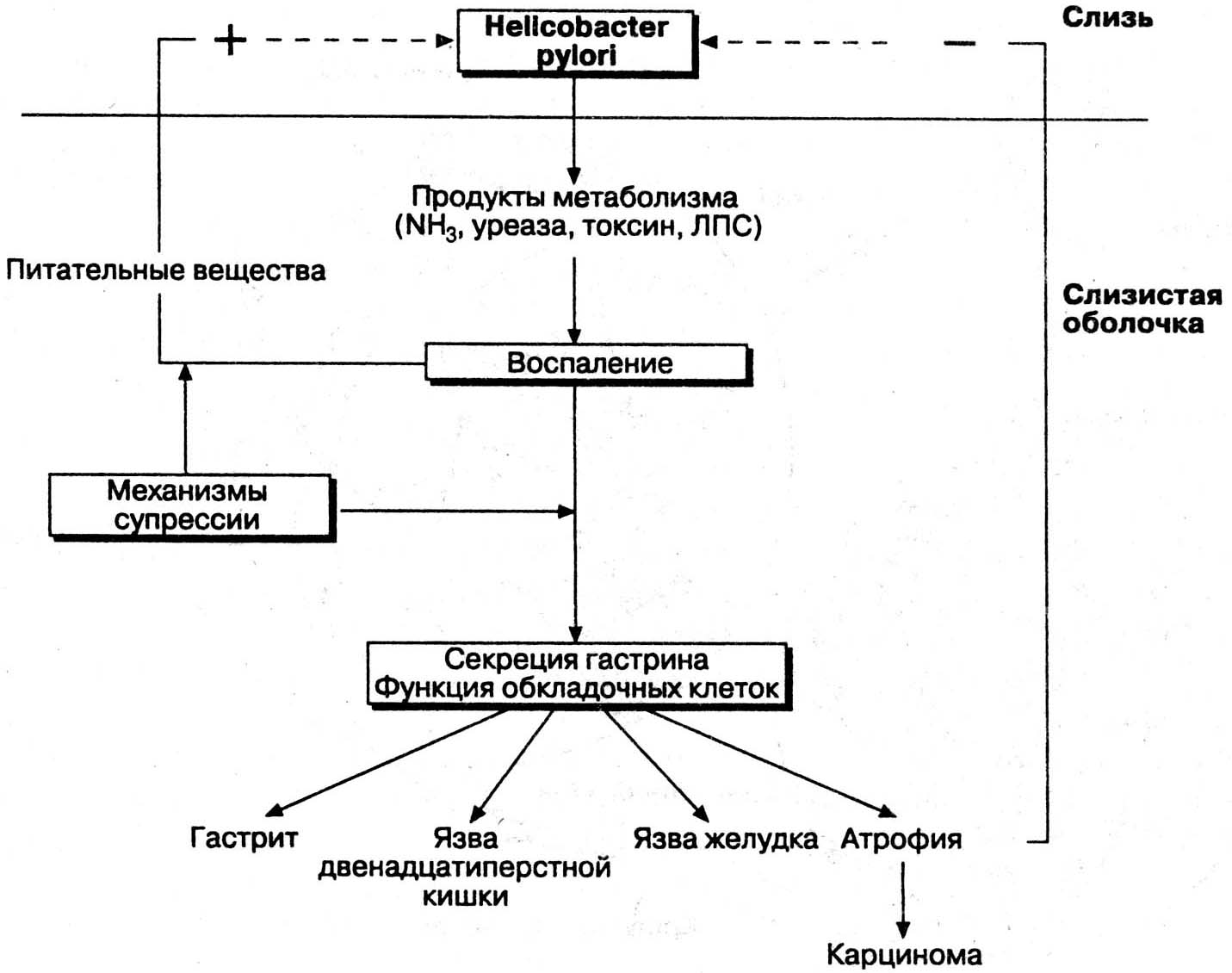
Рис. Возможные механизмы повреждения слизистой оболочки под действием Helicobacter pylori: прямое воздействие (аммиак [NH3], липополисахариды [ЛПС], уреаза, цитотоксин); непрямое (гастрин, соматостатин); индукция воспалительной реакции (гастрит, дуоденальная язва, язва желудка). (По: Blaser M.J. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology, 102: 772, 1992.)
H. pylori образует аммиак из мочевины защелачивает антральный отдел желудка гиперсекреция гастрина стимуляции обкладочных клеток гиперпродукция НСl.
Ряд штаммов H. pylori выделяют цитотоксины, повреждающие слизистую оболочку в пораженную слизистую устремляются лейкоциты (преимущественно нейтрофилы) инфильтрация слизистой воспалительная реакция дегенеративные изменения слизистой на поверхности образуется эрозия, а затем - язва.
Гиперсекреторный синдром, например, при гастриноме.
Гастринома (синдром Золлингера-Эллисона) - опухоль, продуцирующая гастрин, чаще всего локализуется в поджелудочной железе, реже в двенадцатиперстной кишке (60% малигнизируется). Для гастриномы характерно сочетание тяжелых язвенных процессов в желудке с диареей. Локализация язв необычная - они располагаются постбульбарно, в тощей кишке.
Патогенез ее не совсем ясен, поскольку гастрин на кишечную секрецию прямо не влияет. Предполагают, что значительное количество HCl, вырабатывающейся в желудке, попадает в двенадцатиперстную кишку, ингибирует панкреатические ферменты и вызывает изменения желчных кислот.
Прием нестероидных противовоспалительных средств (НПВП).
В механизме действия НПВП важную роль играет ингибирование биосинтеза простагландинов, связанное с угнетением активности циклооксигеназы (ЦОГ). Имеет два изофермента ЦОГ - ЦОГ-1 и ЦОГ-2. Угнетение активности ЦОГ-1 приводит к нарушению синтеза простагландинов в слизистой оболочке желудка, тогда как угнетение активности изофермента ЦОГ-2 определяет противовоспалительный эффект препаратов.
Ингибирование только одной из разновидностей ЦОГ, а именно ЦОГ-2, позволяет избежать повреждений слизистой оболочки желудка, при сохранении противовоспалительного эффекта. В настоящее время созданы препараты (мелоксикам и др.), избирательно угнетающие ЦОГ-2 без повреждения слизистой оболочки желудка.
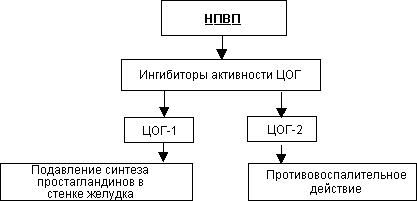
74. – Патогенез язвенной болезни желудка и двенадцатиперстной кишки. Факторы агрессии и защиты. Порочные круги.
В этиологии язвенной болезни также имеют значение другие факторы:
Наследственная предрасположенность (полигенный тип наследования, мультифакториальное заболевание).
Установлен ряд генетических факторов, наличие которых способствует возникновению ЯБ:
наследственно обусловленное увеличение массы обкладочных клеток, их гиперчувствительность к гастрину, повышение образования пепсиногена-1 и расстройство гастродуоденальной моторики могут приводить к повреждению слизистой оболочки желудка и двенадцатиперстной кишки;
Врожденный дефицит фукомукопротеидов слизи, недостаточность выработки секретируемого Ig а и простагандинов снижают резистентность слизистой оболочки;
Группа крови 0 (1), положительный резус-фактор, наличие hla-антигенов в5, в15, в35 и др. Увеличивают вероятность заболевания яб.
Стрессовые факторы (преимущественно тяжелые психические) - существует трактовка язвы как психосоматического заболевания (кортико-висцеральная теория Курцина-Быкова).
Нарушения высшей нервной деятельности под влиянием неблагоприятных экстеро- и интероцептивных сигналов нарушаются взаимоотношения между корой головного мозга и подкорковыми центрами регуляции вегетативных функций повышение секреции HCl, нарушение микроциркуляции в стенке желудка и двенадцатиперстной кишки развитие дистрофических процессов.
Гормоны стресса (прежде всего, глюкокортикоидов) нарушается адгезивность клеток слизистой возникает переваривание слизистой пепсином. Глюкокортикоиды ингибируют синтез простагландинов (они угнетают секрецию HCl в желудке) повышение секреции HCl.
Местные неблагоприятные воздействия на желудок и погрешности в питании (нерегулярный прием пищи, чрезмерно горячая пища, алкоголь, острая пища).
Преобладание тонуса парасимпатической нервной системы.
Гипокинезии.
Решающим звеном в патогенезе яб является дисбаланс между факторами "агрессии" и факторами "защиты" слизистой оболочки желудка и двенадцатиперстной кишки.
Нарушение соотношение факторов агрессии и защиты может быть различным в каждом конкретном случае. Так, при ЯБ двенадцатиперстной кишки чаще ведущую роль играет повышение кислотно-пептической агрессии, тогда как при ЯБ желудка на первый план может выступать снижение факторов защиты слизистой желудка и дуоденогастральный рефлюкс.
Рис. Основные патогенетические механизмы язвообразования
Факторы агрессии
Усиление воздействия ацидо-пептического фактора, связанное с увеличением продукции соляной кислоты (Шварц, 1910 г.: «без кислоты нет язвы») и пепсина.
Нарушение моторно-эвакуаторной функции желудка и двенадцатиперстной кишки (задержка или ускорение эвакуации кислого содержимого из желудка, дуоденогастральный рефлюкс). Липаза и желчь, в состав которой входят гипертонические растворы различных химических соединений и олеиновая кислота оказывают повреждающее действие на слизистую желудка при частых рефлюксах желчи в антральный отдел желудка.
Экзогенные химические вещества, обладающие прямым ульцерогенным действием: НПВП, этанол (особенно если его концентрация составляет 10% и более от общего объема содержимого желудка), никотин повреждает микроциркуляторное русло слизистой, подавляет секрецию поджелудочной железой бикарбонатов.
Инфекция гастродуоденальной слизистой оболочки бактерией H. pylori.
Газовый и негазовый ацидоз (хронические обструктивные заболевания легких, эмфизема легких, цирроз печени и почечная недостаточность).
Гормоны АПУД-системы (гистамин, гастрин, кишечные гормоны).
Факторы защиты
Защитные функции слизи.
концентрация и удержание НСО3- у поверхности париетальных клеток;
предотвращение контакта клеток покровного эпителия с пепсином и желудочной липазой;
обволакивание только что проглоченных частичек пищи, смазывание покрова желудка, т.е. предохранение его от повреждений грубой пищей;
частичная нейтрализация Н+ с помощью отрицательно заряженных гликопротеинов и пептидов слизи;
захват бактерий поступившие в просвет желудка и двенадцатиперстной кишки.
Защитный слой эпителиальных клеток и его активная регенерация (препятствует - катаболическое действие глюкокортикоидов при стрессе).
Секреция бикарбонатов в желудке. Внутриклеточная карбоангидраза превращает СО2 и Н2О в НСО3- и H+. NaНСО3 поступает в клетки с током крови из интерстициальной ткани, добавляя буферные вещества к внутриклеточному пулу бикарбонатов. Через НСО3-/Cl--ионоoбменники апикальной мембраны происходит выделение НСО3- в неактивный слой слизи.
Микроциркуляция крови в слизистой оболочке (возможны острые стрессорные язвы желудка при кровопотерях).
Цитопротекция - способность простагландинов предотвращать или смягчать повреждения слизистой желудка (стимулируют продукцию НСО3- и слизи, усиливают кровоток в слизистой оболочке, подавляют обратную диффузию ионов Н+ из просвета желудка в клетки эпителия и стимулируют восстановление поверхностного эпителия).
Секретин, соматостатин.
Иммунная защита.
75. – Этиология и патогенез панкриотитов. Расстройства кишечной секреции и моторики.
Воспалительные процессы в ткани поджелудочной железы приводят к развитию панкреатита, который может быть острым и хроническим.
Острый панкреатит
В этиологии острого панкреатита существенное значение придают:
злоупотреблению алкоголем,
перееданию жирной пищи,
желчным камням и полипам просвета поджелудочной железы,
механическому повреждению,
инфекциям и интоксикациям.
Наиболее разработанной гипотезой патогенеза острого панкреатита является теория самопереваривания.
В результате действия этиологических факторов происходит повышение секреции панкреатического сока и нарушение его оттока из поджелудочной железы.
Согласно ей протеолитические ферменты поджелудочной железы (трипсиноген, химотрипсиноген, проэластаза и фосфолипаза А2) активируются не в кишечнике, а в поджелудочной железе под воздействием этиологических факторов острого панкреатита активированные ферменты (особенно трипсин) переваривают собственные ткани железы отек, интерстициальные кровотечения, повреждение сосудов, некрозу паренхиматозных клеток.
Активируются и высвобождаются брадикининовые пептиды и другие сосудоактивные вещества (гистамин, серотонин) выраженные нарушения в микроциркуляторном русле не только железы, но и микроциркуляции всего организма. В конце концов может развиться некротизирующий панкреатит. В тяжелых случаях развивается панкреатический шок.
Хронический панкреатит
Основные этиологические факторы действуют или непосредственно на ацинарные клетки железы, вызывая их повреждение и некроз (например, алкоголь или лекарства), или способствуют повышению давления в протоках железы (желчно-каменная болезнь, дуоденальные факторы). Все это приводит к внутрипанкреатической активации протеолитических ферментов железы, аутолизу клеток железы, активации кининовой системы и нарушению процессов микроциркуляции, отеку интерстициальной ткани железы. Хронизации воспалительного процесса способствуют иммунологические нарушения, нарушения антигенного состава клеток железы и включение патологических иммунных реакций. У больных в фазы обострения процесса наблюдается феномен "уклонения" ферментов в общий кровоток, что приводит в свою очередь к ряду системных нарушений.
76. – Печеночная недостаточность. Патогенетические варианты. Нарушение обмена веществ при печеночной недостаточности.
Печеночная недостаточность (пн) – синдром, характеризующийся снижением одной, нескольких или всех функций печени ниже уровня, необходимого для обеспечения нормальной жизнедеятельности организма.
ПН делится на виды по следующим признакам:
По числу нарушенных функций:
парциальная;
тотальная.
По течению:
острая;
хроническая.
По исходу:
летальная;
нелетальная.
Причины печеночной недостаточности
Патологические процессы, локализующиеся в печени и желчевыводящих путях:
гепатиты: вирусные, бактериальные, токсогенные;
дистрофии (гепатозы);
циррозы;
опухоли печени;
паразитарные поражения ее;
генетические дефекты гепатоцитов;
камни, опухоли, воспаления желчевыводящих путей с выраженным холестазом.
Внепеченочные патологические процессы:
шок, в том числе послеоперационный;
сердечная недостаточность;
общая гипоксия;
почечная недостаточность;
белковое голодание;
гипоавитаминоз Е;
дефицит селена;
эндокринопатии, в частности острая недостаточность надпочечников.
Патогенез печеночной недостаточности
Действие этиологического фактора изменение молекулярной архитектоники мембран гепатоцитов усиление свободнорадикального и перекисного окисления липидов частичная или полная деструкция мембран + повышение их проницаемости выход из лизосом их гидролаз, что потенцирует повреждение мембран клеток освобождение поврежденными макрофагами фактора некроза опухоли и интерлейкина-1, способствующих развитию воспалительной и иммунной реакции в печени образование аутоантител и аутосенсибилизированных Т-киллеров, вызывающих дополнительное аутоаллергическое повреждение гепатоцитов.
Нарушение участия печени в углеводном обмене заключается в:
гипогликемия натощак из-за снижения способности гепатоцитов расщеплять гликоген до глюкозы и снижения активности инсулиназы;
гипергликемия после приема пищи из-за снижения способности гепатоцитов превращать глюкозу в гликоген.
Это обусловливает характерный признак печеночной недостаточности (ПН) - неустойчивый уровень глюкозы крови.
Нарушение участия печени в липидном обмене характеризуется снижением способности гепатоцитов:
превращать более атерогенную форму холестерина (свободный холестерин) в менее атерогенный эстерифицированный холестерин;
образовывать липопротеиды, обладающие антиатерогенным действием (ЛПВП (-липопротеипды)).
образование холестериновых и пигментных камней: черные – билирубинат кальция, коричневые – билирубинат кальция + жирные кислоты (холевая, дезоксихолевая, таурохолевая).
Первые два изменения ведут к увеличению в крови уровня свободного холестерина и к снижению антиатерогенных фосфолипидов, что способствует отложению холестерина в стенках сосудов и развитию атеросклероза.
Нарушение участия печени в белковом обмене включает:
снижение синтеза гепатоцитами альбуминов гипоальбуминемия и гипоонкия крови, что на стадии развития портальной гипертензии способствует развитию асцита;
диспротеинемия - снижение количества альбуминов и увеличение содержания в плазме глобулинов.
уменьшение биосинтеза ферментов и белков - прокоагулянтов (протромбина, проакцелерина, проконвертина) развитие коагулопатий (этому способствует также уменьшение всасывания в кишечнике жирорастворимого витамина К, поскольку ПН сочетается с нарушением желчеобразовательной и желчевыделительной функции печени);
снижение активности процесса дезаминирования аминокислот и синтеза мочевины из аминогрупп и аммиака в орнитиновом цикле, что ведет к снижению в крови содержания мочевины. Дефицит -кетоглутаровой к-ты снижение окисления и образования АТФ.
Нарушение биосинтеза гепатоцитами ферментов заключается в уменьшении секреции гепатоцитами в кровь образуемых ими ферментов (холинэстеразы, инсулиназы и др.). Кроме того, повреждение гепатоцитов сопровождается увеличением выхода из них в кровь внутриклеточных ферментов: аланин-трансаминазы и глютамат-трансаминазы.
Расстройство обмена витаминов состоит в:
снижении всасывания в кишечнике жирорастворимых витаминов A, D, Е, К;
уменьшении способности гепатоцитов превращать провитамины в активные витамины (например, каротин в витамин А);
торможении процесса образования из витаминов коферментов (например, из пантотеновой кислоты - ацетил коэнзим А, из витамина B1 - кокарбоксилазы пирувата).
77. – Виды, механизмы возникновения и дифференциальная диагностика желтух.
Желтухи
Подпеченочная (обтурационная, механическая) желтуха.
камни в желчевыводящих путях;
воспаление желчевыводящих путей;
паразиты в желчном пузыре;
дискинезия желчевыводящих путей;
опухоли, в т.ч головки поджелудочной железы.
Стойкое нарушение выделения желчи из желчных капилляров, желчного пузыря или его протока в просвет двенадцатиперстной кишки желчь и находящийся в ней связанный билирубин-диглюкуронид (БДГ) задерживаются и поступают обратно в желчные капилляры, а оттуда в гепатоциты и кровь в крови повышается содержание БДГ в силу растворимости его в воде проходит через почечный фильтр БДГ выделяется с мочой.
В кишечник желчь не попадает, уробилиноген и стеркобилиноген не образуются.
Таким образом, при подпеченочной желтухе содержание билирубина в крови повышается более 20 ммоль/л за счет фракции связанного прямого билирубина, в моче определяется билирубин, в кале нет стеркобилина (кал обесцвечен).
Надпеченочная (гемолитическая) желтуха
Причины (см. раздел «Гемолитические анемии»).
Гемолиз в РЭС повышен распад эритроцитов увеличивается количество свободного билирубина, его содержание в крови повышается повышенное количество поступает в печень образуется больше, чем в норме связанного билирубина (БДГ) БДГ в большем количестве поступает в желчные пути, а оттуда в кишечника в большем количестве образуется уробилиноген (УБ) и стеркобилиноген (СБ) УБ в большем количестве поступает в печень, оттуда в кровь и выделяется с мочой, в кале повышается содержание стеркобилина.
Таким образом, при надпеченочной желтухе в крови повышается общее содержание билирубина более 20 ммоль/л за счет фракции свободного неконъюгированного билирубина, в моче определяется уробилин, в кале повышается содержание стеркобилина.
Нарушение внутрипеченочного кровотока при сердечной недостаточности или при портокавальном шунтировании также нарушает доставку билирубина к гепатоцитам и приводит к небольшой неконъюгированной гипербилирубинемии.
Печеночная (паренхиматозная) желтуха
Возникает вследствие нарушений захвата, связывания и экскреции билирубина. Часто эти механизмы комбинируются, но иногда проявляются изолированно.
Клиника и лабораторные признаки желтухи во многом еще зависят от фазы патологического процесса - обострение, ремиссия.
В фазе обострения происходят некротические процессы в печени повышается проницаемость мембран гепатоцитов, происходит отек и нарушение функции желчных капилляров снижение захвата Бсв., обратное поступлению в кровь БДГ выделение с мочой БДГ и уробилина (из-за неспособности печени разрушать уробилиноген до ди-трипирролов), снижение стеркобилина в кале.
По лабораторным признакам в это время паренхиматозная желтуха похожа на механическую. По мере стихания обострения в патогенезе большую роль имеет дефект захвата билирубина и лабораторные признаки "похожи" на надпеченочную желтуху.
Таким образом, для паренхиматозной желтухи характерно повышение билирубина сыворотки за счет свободного и связанного, появление в моче билирубина и уробилина, снижение стеркобилина в кале.
Нарушение захвата билирубина гепатоцитами в синусоидах может наблюдаться при синдроме Жильбера и в ответ на прием некоторых препаратов, например рифампицина.
Снижение функции УДФ-глюкуронилтрансферазы также может быть причиной нарушения конъюгации билирубина у новорожденных при синдроме Жильбера и синдроме Криглера-Найяра I и II типов.
Дифференциальная диагностика различных типов желтух
|
Гемолитическая (надпеченочная) |
Паренхиматозная (печеночная) |
Механическая (подпеченочная) |
Прямой билирубин крови |
норма |
повышен |
повышен |
Непрямой билирубин крови |
повышен |
повышен |
повышен |
Билирубиноген мочи |
не определяется |
определяется |
определяется |
Уробилиноген мочи |
понижен |
повышен |
понижен |
Стеркобилин кала |
повышен |
понижен |
понижен |
78. – Энзимопатии при патологии печени. Ферменты-маркеры повреждения гепатоцитов и холестерина.
Для диагностики холестаза используют энзиматические тесты. К ферментам – маркерам холестаза относят щелочную фосфатазу, лейцинаминопептидазу и гаммаглутамилтрансферазу, которые синтезируются на внешней поверхности каникулярной мембраны гепатоцитов.
Сывороточные энзимные тесты оценки холестаза
Ферменты |
Референтные пределы |
Щелочная фосфатаза (ЩФ) |
1,63 – 4,65 мккат/л |
Гаммаглутамилтрансфераза (ГГТ) |
0,20 – 0,60 мккат/л |
Лейцинаминопептидаза (ЛАП) |
1,90 – 4,00 мккат/л |
Наиболее достоверным признаком холестаза является повышение в сыворотке активности экскреторного фермента щелочной фосфатазы. Если повышена активность в сыворотке только щелочной фосфатазы, то при отсутствии костной патологии это самый ранний показатель возможного обширного вовлечения печени в патологический процесс. В ранние сроки внутрипеченочного холестаза повышение активности щелочной фосфатазы является следствием активации ее синтеза, далее ее увеличение, особенно в форме макрощелочной фосфатазы, связано с деструкцией желчных канальцев при действии желчных кислот.
Увеличение активности гаммаглутамилтрансферазы чаще всего наблюдается при повреждении печени при хроническом алкоголизме.
Увеличение активности всех трех ферментов – признак гепатобилиарного поражения.
Синдром цитолиза (повреждения гепатоцитов)
Энзимные тесты, являются высокочувствительные индикаторами цитолиза гепатоцитов. Определение активности АлАТ, например, характеризуется как «безпункционная биопсия печени».
Сывороточные энзимные тесты оценки повреждения гепатоцитов
Ферменты |
Референтные пределы |
Аланинаминотрансфераза (АлАТ) |
0,13 – 0,19 мкмоль/л |
Аспартатаминотрансфераза (АсАТ) |
0,03 – 0,13 мкмоль/л |
Лактатдегидрогеназа (ЛДГ) |
1,90 – 4,60 мкмоль/л |
Наиболее информативным маркером цитолиза гепатоцитов является АлАТ. Это обьясняется тем, что АлАТ содержится исключительно в гиалоплазме гепатоцитов, а АсАТ включает изоэнзим, локализующийся в митохондриях. Поэтому при цитолитическом процессе, развивающемся в печени, преобладает вымывание АлАТ. С другой стороны, при преобладании некробиоза клеток с вовлечением в процесс митохондрий, например, в клетках сердечной мышцы при остром инфаркте миокарда, превалирует элиминация АсАТ. Поэтому нередко АлАТ называют «печеночной», а АсАТ - «сердечной» трансаминазой. Параллельное определение двух энзимов позволяет полнее охарактеризовать происхождение гиперферментемии.
Обычно рассчитывается коэффициент де Ритиса - АсАТ/АлАТ, в норме близкий к 1. Его снижение до 0.7 дополнительно подтверждает «печеночный» характер, а повышение до 1,3 и более - «непеченочный» генез гиперферментемии. Значительное снижение коэффициента (<0.7) рассматривают как индикатор тяжелого повреждения печени.
Лактатдегидрогеназа (ЛДГ) содержится в сердечной и скелетной мышцах, печени, легких. Повышение содержания ЛДГ типично для гепатоцеллюлярных заболеваний и менее типично для холестатических. Изофермент ЛДГ5 - содержится в печени.
79. – Этиология гепатитов. Патогенез цирроза печени. Этиология и патогенез портальной гипертензии.
Гепатиты
Острые гепатиты.
Хронические гепатиты (6 мес. и более) - нарастающая пролиферация мезенхимальных клеток, фиброз портальных полей, коллагеноз внутридольковой стромы, перипортальная и внутридольковая мононуклеарная инфильтрация, некроз и некробиоз гепатоцитов.
Виды хронических гепатитов: аутоиммунный, вирусный, лекарственный, билиарный, первичный склерозирующий холангиит.
Циррозы печени - дистрофия клеток вплоть до некроза, дезорганизация стромы долек соединительно-тканными тяжами, узловая регенерация с функционально неполноценными гепатоцитами, диффузное развитие соединительной ткани (фиброз), портальная гипертензия, печеночно-клеточная недостаточность
Крупноузловой (постнекротический).
Мелкоузловой (портальный).
Билиарный (холестатический):
внутрипеченочный – нарушение образования желчи на уровне гепатоцита;
внеипеченочный – органические нарушения секреции и оттока желчи.
Портальная гипертензия – синдром, сопровождающийся повышением давления в бассейне воротной вены, связанный с наличием препятствия оттоку крови (блок).
В зависимости от локализации блока выделяют следующие формы портальной гипертензии:
Постгепатическая (надпеченочная) связана с препятствием во внеорганных отделах печеночных вен или в нижней полой вене проксимальнее места впадения в нее печеночной вены (синдром Бада-Киари, врожденное мембранозное заращение нижней полой вены, первичные сосудистые опухоли (лейомиома и др.), повышение давление в нижней полой вене при правожелудочковой сердечной недостаточности).
Внутрипеченочная связана с блоком в самой печени:
постсинусоидальный блок (циррозы печени, хронический алкогольный гепатит, веноокклюзионная болезнь и т.д.);
парасинусоидальный блок (хронический гепатит, массивная жировая инфильтрация печени);
пресинусоидальный блок (гепатоцеребральная дистрофия, первичный билиарный цирроз, метостазы печени и др.);
Прегепатическая (подпеченочную) связана с препятствием в стволе воротной вены или ее крупных ветвей (тромбоз воротной вены, сдавление воротной вены опухолью и т.д.).
Основным патогенетическим фактором синдрома портальной гипертензии является механическое препятствие оттоку крови.
Наиболее характерное следствие портальной гипертензии - образование коллатералей между бассейном воротной вены и системным кровотоком.
При прегепатической портальной гипертензии развиваются портальные анастомозы, восстанавливающие ток крови из отделов портальной системы, расположенных ниже блока, во внутрипеченочные ветви портальной системы.
При внутри- и надпеченочной портальной гипертензии анастомозы обеспечивают отток крови из системы воротной вены в обход печени в бассейн верхней или нижней полой вены.
Схема венозной системы печени
|
На рисунке представлена схема венозной системы печени: 1 - v.portae, 2 - v.lienalis, 3 - v.mesenterica superior, 4 - v.v.hepaticae. |
Она складывается из приводящих и отводящих вен. Приводящие вены - это система воротной вены, которая формируется из двух стволов - селезеночной и верхней брыжеечной вены.
Войдя в ворота печени вместе с ветвями печеночной артерии, венозные разветвления воротной вены образуют капиллярную сеть, которая несет смешанную артериальную и венозную кровь. В синусоидах кровь контактирует с гепатоцитами. Отводящими коллекторами являются печеночные вены, которые впадают в нижнюю полую вену.
Нарастающее гидростатическое давление в портальной системе наряду с повышенным притоком через печеночную артерию постепенно приводит к повышению давления в основном стволе воротной вены примерно. Пока это происходит, развивается интенсивная коллатеральная циркуляция, которая на первых порах компенсирует повышение давления в системе воротной вены. Эти коллатеральные анастомозы существую и в норме, но они не приспособлены к такому повышению кровотока, венозные стволы варикозно расширяются.
Особенно опасны коллатерали в области пищевода, поскольку могут развиваться тяжелые кровотечения. Расширяются также коллатеральные анастомозы в области передней брюшной стенки, а также в области прямой кишки.
Шунтирование крови в обход печеночной паренхимы по сути означает частичное функциональное отключение печени, последствия которого для организма весьма многообразны. К важнейшим из них относятся:
Бактериемия - результат выключения РЭС печени, что обусловливает повышенный риск "метастатической" инфекции.
Эндотоксемия.
Гиперантигенемия - перегрузка антигенным материалом кишечного происхождения иммунной системы организма.
С повышением давления в воротной вене связано образование асцита. Чаще он встречается при постгепатической и внутригепатической портальной гипертензии.
80. – Патогенез печеночной комы.
Печеночная кома (от греч. «koma» - глубокий сон) развивается как финальный этап нарастающей по тяжести тотальной печеночной недостаточности. В комах погибают 80% больных.
Выделяют два варианта механизма развития печеночной комы: шунтовой и печеночно-клеточный.
Шунтовая кома обусловлена поступлением в кровоток и интоксикацией ЦНС продуктами гниения белков из кишечника (индола, скатола, фенола, путресцина, кадаверина и др. печеночный запах), которые в норме обезвреживаются в печени и выделяются почками. Возникает при портальной гипертензии (например, при циррозе печени) высокое гидростатическое давление в портальной системе сброс крови по шунтам в нижнюю полую вену в общий кровоток.
Пища, богатая белком, повышает вероятность развития комы из-за всасывания токсичных продуктов распада белков, попадающих в общий кровоток. Среди них аммиак, карбаминово-кислый аммоний, путресцин, кадаверин, метионин и др.
Гепато-целлюлярная (печеночно-клеточная) кома возникает при массивном некрозе паренхимы печени, когда существенно снижаются ее гомеостатическая и барьерная функции. В основе развития комы лежат несколько взаимосвязанных патогенетических механизмов.
Гипогликемия.
Тяжелый ацидоз: в крови нарушается щелочно-кислотное равновесие, нарушается соотношение молочной и пировиноградной кислот сдвиг рН в кислую сторону внутриклеточный ацидоз отек мозга и печени. Ацидоз также способствует входу калия и внутриклеточной гипокалиемии.
Интоксикация организма, обусловленная появлением и нарастанием в крови уровня веществ, оказывающих общетоксическое и, церебротоксическое действие.
в крови увеличивается уровень свободного аммиака, что обусловлено нарушением преобразования его в мочевину в орнитиновом цикле гепатоцитов избыток аммиака повреждает клетки органов и тканей, угнетает ферментные реакции в них и нарушат работу энзимов цикла трикарбоновых кислот (большая часть -кетоглютаровой кислоты используется на связывание избытка аммиака с образованием глутаминовой кислоты дефицит -кетоглютарата резко снижает интенсивность процессов окисления, сопряжения окисления и фосфорилирования дефцит АТФ.
поврежденные гепатоциты подвергаются деструкции содержащиеся в них вещества попадают в кровь и оказывают патогенное действие на клетки органов и тканей, в том числе - нервной системы.
в крови нарастает содержание не захватываемого печенью свободного билирубина, токсически действующего на клеточные мембраны.
нарастает поступление в кровь высокотоксичных продуктов распада ароматических аминокислот (индола, скатола, фенола), а также гнилостного разложения белков (путресцина, кадаверина).
Нарушается системная гемодинамика (из-за общей интоксикации организма): сердечный выброс, АД, ОЦК.
Нарушения гемостаза (дефицит протромбина, фибриногена и других факторов, изменение ее реологических свойств) кровотечений, кровоизлияния, сладжирование крови в микрососудах органов и тканей.
Возникает прогрессирующая общая гипоксия смешанного характера.
81. – Этиология и патогенез острой печеночной недостаточности.
Почечная недостаточность – это синдром, развивающийся в результате тяжелых нарушений почечных процессов, приводящих к расстройству гомеостаза, и характеризующийся азотемией, нарушением водно-электролитного состава и кислотно-щелочного состояния организма.
Острая почечная недостаточность (ОПН) может возникнуть внезапно вследствие острых, чаще всего обратимых заболеваний почек (старый термин «уремия», поскольку в основном ориентируется на нарушение выделения шлаков – остаточный азот, мочевина).
Этиология опн
Преренальная ОПН.
гемодинамические нарушения с гиповолемией и снижением клубочковой фильтрации (шок, тромбоз, эмболия, сепсис, ожоги, рвота, понос); ОПН развивается при снижении фильтрации до 30%;
снижение клубочковой фильтрации без гиповолемии (дефицит воды и соли, острая гиперкальциемия, лекарственный норадреналиновый спазм почечных сосудов).
Ренальная ОПН.
острый тубулярный или кортикальный некроз (нефротоксические вещества: CCl4, неорганические соединения ртути, тяжелые металлы, этиленгликоль, бледная поганка, антибиотики (гентамицин, неомицин), фенацетин, сульфаниламиды; острые гемолиз и миоли: краш-синдром, гемолитическая анемия, гемотрансфузионные осложнения; гломерулонефриты (редко); шок с некоррегируемой преренальной ОПН);
блокада канальцев (уратами, сульфаниламидами).
Постренальная ОПН (почечные камни, опухоли тазовых органов, в т.ч. аденома простаты, острый пиелонефрит).
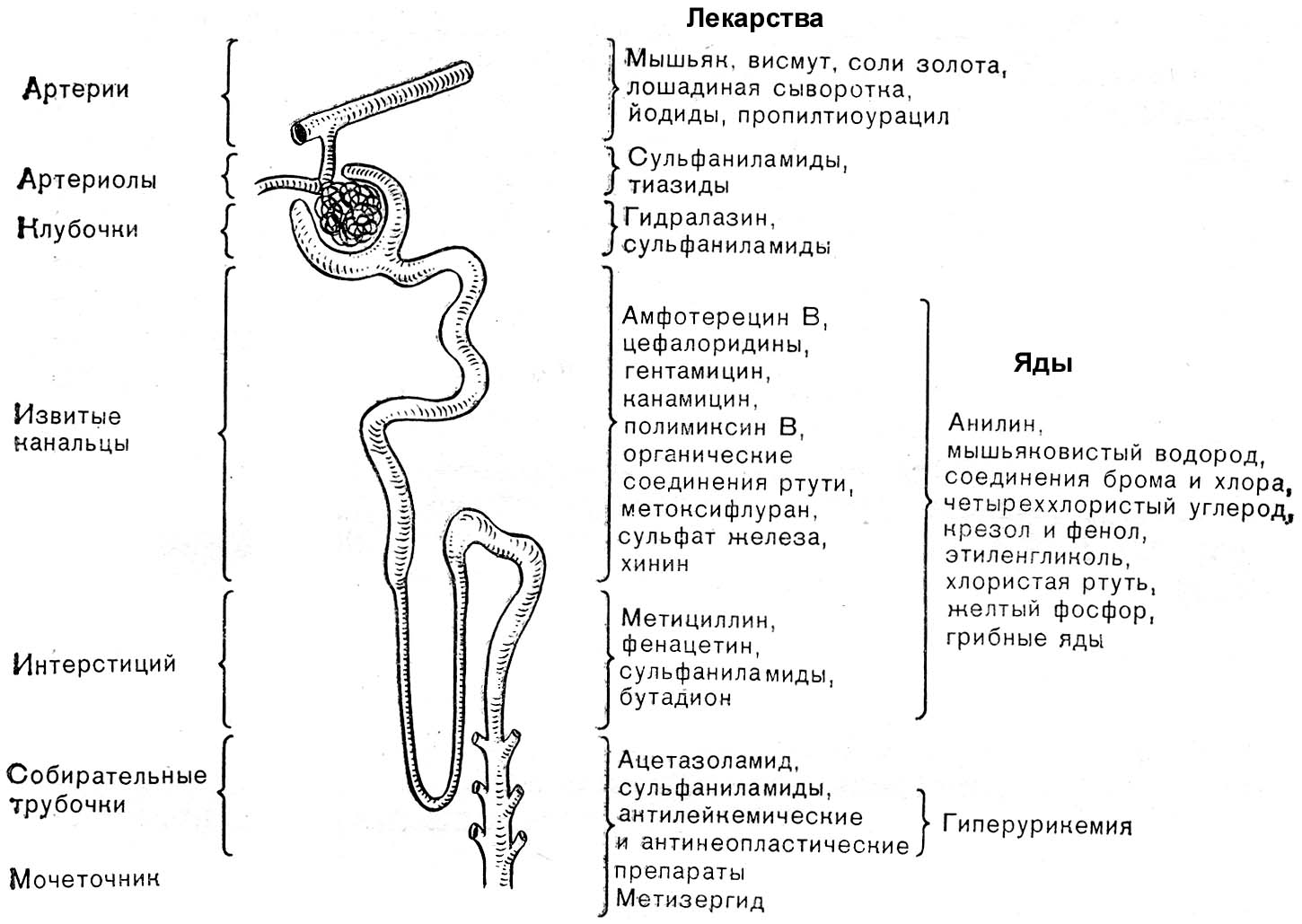
Рис. Место действия на почку важнейших нефротоксинов.
Основные звенья патогенеза опн
Значительное снижение объема клубочковой фильтрации вследствие:
гипоперфузии клубочков в результате ишемии почек «преренального» генеза («критическим» считают уровень давления крови в афферентных артериолах, равный 40-60 мм рт.ст.);
вазоконстрикции почечных артериол, развивающейся в связи с гипотензией и гипоперфузией почек;
микротромбоза и(или) агрегации клеток крови в микрососудах почек (при различных видах шока, сопровождающихся образованием избытка факторов коагуляции крови, - септическом, травматическом, геморрагическом).
Сужение или обтурация большого числа канальцев почек в результате:
накопления в поврежденных клетках гидрофильных ионов кальция отека и набухания эпителия, что уменьшает просвет канальцев;
закрытия их клеточным детритом (повреждение и гибель эпителия) или цилиндрами, состоящими из белка (при развитии воспаления или повышении проницаемости клубочкового фильтра), миоглобина (у пациентов с травмами мышц), гемоглобина (у больных с гемолизом эритроцитов);
Подавление процессов экскреции и секреции, протекающих в эпителии канальцев под действием нефротоксических факторов.
Дополнительное (к действию названных выше механизмов) повреждение клубочков, канальцев, интерстициальной ткани в связи с развитием воспалительной и иммуноаллергической реакций в ответ на прямое повреждение указанных структур (этот механизм нередко обусловливает «переход» ОПН в ХПН).
Стадии опн
Шоковая (начальная) стадия длится несколько часов - 3 дня, преобладают симптомы основного заболевания + нарушение гемодинамики с выраженной гипотензией («циркуляторный коллапс»).
Олиго-анурическая стадия длится 7-10 дней, олигурия менее 500 мл/сут., характерны сонливость, тошнота, рвота, судороги, гипергидратация, в крови нарастает содержание мочевины, креатинина, и др. продуктов азотистого обмена, калия, магния, сульфатов, фосфатов, ацидоз; шлаки выделяются в серозные полости ( шумы трения), слюнные, желудочные, кишечные железы ( мочевой запах изо рта).
Восстановление диуреза (полиурическая стадия) продолжается около 3 недель, появляется опасность больших потерь жидкости и электролитов, легкого присоединения инфекции.
Выздоровление.
82. - Этиология и патогенез хронической печеночной недостаточности.
Хроническая почечная недостаточность (ХПН) - синдром, развивающийся в связи с нарастающей гибелью и значительным уменьшением количества функционирующих нефронов, характеризующийся постоянным прогрессирующим нарушением функции почек.
Этиология хпн
«Пререналъные» причины (хронические артериальные гипертензии, медленно прогрессирующий стеноз почечных артерий).
«Ренальные» причины (хронические заболевания почек: гломерулонефриты, пиелонефриты, поликистоз, туберкулез, опухоли, амилоидоз и др.; патология других органов, сопровождающаяся вторичным поражением почек, - сахарный диабет, системная красная волчанка, диспротеинозы и др.; воздействие тяжелых металлов, лекарственных веществ: панадол, парацетамол (анилиновые) фенацетиновый нефрит, сульфаниламиды сульфаниламидная почка, стрептомицин, неомицин, канамицин (антибиотики-аминогликозиды)).
«Постреналъные» причины (обструкция мочевыводящих путей при мочекаменной болезни, аденоме простаты).
Стадии хпн
Полиурическая стадия – нарушение концентрирующей функции почек ( реабсорбции, диурез до 150 л/сут.).
Олигурическая стадия – нарушение фильтрационной функции (диурез менее 500 мл).
Терминальная стадия.
Патогенез хпн
Патогенез сводится к прогрессирующему снижению, вплоть до прекращения, процессов клубочковой фильтрации плазмы крови, канальцевой экскреции, секреции и реабсорбции в связи с нарастающей гибелью нефронов и замещением их соединительной тканью.
Накопление азотистых шлаков (увеличение уровня остаточного азота)
Остаточный азот:
Мочевина нетоксична, но возможна дегидратация клеток мозга и синтез гуанидина.
Аммиак токсичен, механизм токсического действия обусловлен связыванием его с -кетоглютаровой кислотой.
Мочевая кислота способствует образованию почечных камней (дефицит уромокоида), развитию подагры.
Креатинин и креатин нетоксичны, диагностическое значение имеет креатинин.
Азтистые шлаки выделяются кожей и слизистыми кожный зуд, перикардит, плеврит (шум трения плевры и перикарда), абдоминальные боли (поражение брюшины), поносы, стоматиты, мочевой запах (из-за аммиака), уремическое легкое.
Желтоватый цвет кожи (из-за накопления в ней урохрома).
Интоксикация ЦНС слабость, повышенная утомляемость, потеря интереса к окружающему, потеря памяти, ухудшение сна, головные боли, дыхание Чейн-Стокса, Куссмауля (протоны возбуждают дыхательный центр).
Токсическое поражение миокарда миокардиты.
Токсическое поражение печени нарушение всех функций печени.
Токсическое поражение эндокринных органов.
Нарушение всасывания в ЖКТ.
Интоксикация иммунокомпетентных органов.
Анемия обусловлена:
синтеза эритропоэтина в почке;
деффицит железа (нарушение всасывания, синтеза апоферрина, трансферрина, ионизации в желудке);
миелотоксикоз (эритропения и тромбоцитопения);
кровоточивость ( синтеза факторов свертывания, ломкость капилляров, тромбоцитопения и тромбоцитопатия);
повышенный эритродиерез (дефектные эритроциты с малой продолжительностью жизни).
Нарушение водно-электролитного баланса
Нарушение фильтрации + альдостеронизм (альдостерон не инактивируется печени) задержка Na+ гипернатриемия (особенно в олигурическую стадию).
В полиуричекую стадию реабсорбции К+ + альдостеронизм гипокалиемия.
Утечка К+ из поврежденных клеток + нарушение его секреции (Н+ К+ в эритроцитах) гиперкалиемия.
Гипокальциемия вследствие:
повышенная потеря с мочой кальция остеопороз;
вторичный гиперпаратиреоидизм вымывание кальция из костей и гиперфосфатемия остеопороз, кариес, парадонтоз.
нарушение превращения прогормона витамина D3 в активный гормон - витамин D3 образования кальцийсвязывающего белка в кишечнике, реабсорбции кальция в почечных канальцах.
изменение водного обмена: сначала дегидратация вследствие полиурии, затем гипергидратация отеки (см. патогенез ниже), левожелудочковая сердечная недостаточность.
Изменение кислотно-щелочного равновесия:
ацидоз (способствует активации остекластов (Са++ Н+):
гипоксия сложного генеза;
аммиака;
страдают механизмы почечного ацидогенеза;
бикарбонатов плазмы.
в терминальной стадии присоединяется рвота, понос потеря натрия и хлоридов возникает гипохлоремический алкалоз.
83. – Патогенез почечных отеков.
Почечные отеки условно делят на нефритические и нефротические (евозможен нефрит без вторичного (из-за нарушения кровотока) повреждения канальцев.
Нефритический отек (воспаление клубочков)
Основную роль при поражении клубочкового аппарата почки играет активация ренин-ангиотензин-альдостероновой системы, следствием которой является задержка натрия и воды. Кроме этого определенную роль играет также повышение проницаемости клубочков из-за их иммунокомплексного повреждения (при гломерулонефрите, СКВ) или токсического повреждения.
Роль гиалуронидазы. Гиалуронидаза («фактор проницаемости») является ферментом микробного или тканевого происхождения, обладающим специфическим свойством - вызывать деполимеризацию и гидролиз гиалуроновой кислоты (входящей в состав межклеточного вещества стенки сосудов и капилляров). При некоторых заболеваниях ее активность в сыворотке крови возрастает (у больных с острым и обострением хронического гломерулонефрита, с нефротическим синдромом). Это вызывает деполимеризацию гиалуроновой кислоты, и сопровождается увеличением размеров микропор в сосудистой и капиллярной стенках, через которые усиливается уход в ткани не только воды и ионов (в том числе и ионов натрия), но и мелкодисперсных фракций белка (альбуминов). Таким образом, основное вещество соединительной ткани приобретает резко выраженные гидрофильные свойства, что также играет существенную роль в развитии отеков.
Нефротический отек (поражение турбулярного аппарата)
Поражение канальцевого аппарата почки сопровождается гиперпртеинурией, а ее следствием является гипоонкия плазмы из-за альбуминов плазмы. Снижение онкотического давления крови неизбежно приводит к потере ее жидкой части, т.е. развивается гиповолемия раздражение осмо- и волюморецепторов гипоталамуса. Возбуждение осморецепторов – стимул для выброса АДГ, волюморецепторов – альдостерона. Оба гормона способствую задержке воды.
84. – Этиология и патогенез гломерулонефрита.
Острый гломерулонефрит – это заболевание инфекционно-аллергической природы с преимущественным поражением капилляров обеих почек. Чаще болеют в возрасте 12-40 лет, несколько чаще мужчины; заболевание сезонное, чаще возникает в странах с холодным и влажным климатом.
Этиология.
Возбудитель - -гемолитический стрептококк группы А (имеется связь со стрептококковой инфекцией: ангина, одонтогенная инфекция, гаймориты, синуситы и др., а также кожные заболевания: рожа, стрептодермия);
Дефицит Т-супрессоров.
Патогенез. В патогенезе играют роль различные иммунологические нарушения.
Образование обычных антител. Комплекс Ag-At может оседать на базальной мембране клубочков, так как она имеет богатую васкуляризацию, то оседают преимущественно крупные депозиты. Реакция Ag-At разыгрывается на самой базальной мембране, при этом присутствует комплемент, биологические активные вещества: гистамин, гиалуронидаза, могут также страдать капилляры всего организма.
При стрептококковой инфекции стрептококковый антиген может повреждать эндотелий почечных капилляров, базальную мембрану, эпителий почечных канальцев - образуются аутоантитела, возникает реакция Ag-At. Причем в роли антигена выступают поврежденные клетки.
У базальной мембраны почек и стрептококка есть общие антигенные структуры, поэтому нормальные антитела в стрептококке могут повреждать одновременно и базальную мембрану - перекрестная реакция.
Аутоиммунные реакции 2-х типов: цитотоксический и иммунокомплексный.
Хронический гломерулонефрит – это двухстороннее воспалительное заболевание почек иммунного генеза, которое характеризуется неуклонной постепенной гибелью клубочков, сморщиванием почки, постепенным снижением ее функции, развитием артериальной гипертензии и смертью от хронической почечной недостаточности.
Заболеваемость мужчин и женщин одинаковая; встречается во всех странах мира, но чаще в холодных.
Этиология до конца не ясна, у части больных – перенесенный ранее острый гломерулонефрит, другие случаи не ясны. Иногда провоцирующим фактором может быть повторная вакцинация, медикаментозная терапия (например, противоэпилептические средства).
Патогенез. В основе иммунологический механизм. Морфологически в области базальной мембраны находят отложения иммунных комплексов, состоящих из иммуноглобулина и комплемента.
85. – Этиология эндокринопатий. Виды эндокринопатий.
Общие механизмы эндокринных нарушений можно разделить по местоположению первичного повреждения на две большие группы, железистые и внежелезистые эндокринопатии.
Железистые эндокринопатии представляют собой нарушения самих эндокринных желез, когда первичное повреждение сводится к одной из желез внутренней секреции; они занимают самую большую долю из общей численности эндокринопатий.
В соответствии с субординационной структурой внутренней секреции большинства гормонов патогенетически различают следующие виды железистых эндокринопатий:
Первичные эндокринопатии – нарушение гормонообразования является следствием повреждения периферической железы.
Вторичные эндокринопатии – обусловлены повреждением гипофиза.
Третичные эндокринопатии – обусловлены повреждением гипоталамуса.
Все эндокринопатии могут проявляться по гипофункциональному и гиперфункциональному типу.
Причиной первичных гипофункциональных эндокринных нарушений является повреждение периферической железы, вместе с тем нередко наряду с дефицитом периферического гормона наблюдается повышение уровня тропного гормона аденогипофиза (по принципу обратной связи). Например, при болезни Аддисона дефицит кортизола сочетается избытком АКТГ. Только периферическим характером отмечаются гипофункциональные синдромы при повреждениях островков поджелудочной железы или околощитовидных желез.
Причиной первичных гиперфункциональных эндокринопатий обычно является опухоль, отличающаяся высокой секреторной активностью, выходящая из "подчинения регулирования обратной связью". Характерным в этом случае является понижение в крови соответствующего аденогипофизарного тропина.
Вторичные гипофункциональные нарушения редко изолированы (дефицит ТТГ - центральный гипотиреоз, дефицит АКТГ - центральный гипокортицизм), чаще страдают все гипофизарные функции с возникновением картины пангипопитуитаризма. Дифференциально диагностическим признаком по сравнению с первичными гипофункциями является сохранившаяся реакция на соответствующий гипофизарный тропин (правда, длительная нехватка аденогипофизарной стимуляции приводит зачастую к атрофии периферической железы). Уровень эндогенных тропинов в крови низкий.
Вторичные гиперфункциональные эндокринопатии обычно вызваны аденомой аденогипофиза и им присущ характер изолированного гиперпитуитаризма (чаше всего базофильная аденома - болезнь Иценко-Кушинга, несколько реже ацидофильная аденома - гигантизм или акромегалия).
Третичные эндокринопатии чаще всего проявляются в области гонадотропинов (эмоциональная аменорея, случаи преждевременного полового созревания).
Рис. Комплексная схема сложной обратной связи с приведением модулирующих факторов влияния вышестоящих нервных центров.
Эндокринная дисфункция - комбинации гипо- и гиперфункциональных нарушений железы или изменение качества ее секреции.
Типичным примером может служить адреногенитальный синдром, когда первичное понижение синтеза и секреции кортизола по принципу обратной связи увеличивает секрецию АКТГ, который способен повышать продукцию надпочечниковых половых гормонов. Следовательно, имеет место в одной железе гипофункция (глюкокортикоидная) и гиперфункция (половых гормонов).
Внежелезистые формы эндокринопатий. Роль транспортных белков, разрушения в плазме и тканях, рецепторов.
Тканевые (внежелезистые) эндокринопатии не сопровождаются нарушением функции собственно железы, а патология обусловлена тканевой утилизацией или обработкой гормона (вторично, по типу обратной связи, может наступить также нарушение деятельности железы).
Тканевые псевдогиперфункциональные нарушения могут быть связаны с:
повышением тканевого преврашения прогормона в гормон (например, тестостерона в дигидростерон в некоторых случаях гирсугизма);
нарушением инактивации гормона в плазме и печени (например, эстрогенов при заболеваниях печени, что ведет к гинекомастии; альдостерона - к гипертензии; образование циркулирующих антител к инсулину, АКТГ, СТГ);
усиление связывания гормона плазменными белками (инсулин, кортизол, тиреоидные гормоны);
переход одних гормонов в другие (тестостерона в эстрогены - гинекомастия).
Особую группу составляют эктопические эндокринные синдромы: например, злокачественные опухоли легких и печени приобретают способность выделять вазопрессин, АКТГ и др. Собственная железа может зачастую затормаживаться по типу обратной связи.
Тканевые псевдогипофункциональные нарушения могут быть следствием
отсутствия рецепторов в органе-мишени (например, при сахарном диабете II типа, нефрогенном несахарном диабете при отсутствии в почках рецепторов для вазопрессина; псевдогипопаратиреозе - отсутствие в почках рецепторов для паратгормона; при отсутствии рецепторов к тестостерону - тестикулярная феминизация; псевдогипоальдостеронизме, резистентности к кортизолу и др.);
ослабление связывания гормона плазменными белками (инсулина, кортизола, тиреоидных гормонов - в некоторых случаях при поражении печени и снижении синтеза плазменных белков);
блокады образования интермедиарных факторов, реализующих эффект гормона (при нарушении синтеза печеночных соматомединов, опосредующих эффект СТГ возникает карликовость, хотя секреция СТГ нормальна);
медикаментозного происхождения - "разгонка" систем микросомального окисления в печени некоторыми фармацевтическими препаратами приводит к инактивации некоторых гормонов (стероидов).
86. – Патофизиологическая характеристика основных вариантов гипопитуитаризма.
Общие механизмы эндокринных нарушений можно разделить по местоположению первичного повреждения на две большие группы, железистые и внежелезистые эндокринопатии.
Железистые эндокринопатии представляют собой нарушения самих эндокринных желез, когда первичное повреждение сводится к одной из желез внутренней секреции; они занимают самую большую долю из общей численности эндокринопатий.
В соответствии с субординационной структурой внутренней секреции большинства гормонов патогенетически различают следующие виды железистых эндокринопатий:
Первичные эндокринопатии – нарушение гормонообразования является следствием повреждения периферической железы.
Вторичные эндокринопатии – обусловлены повреждением гипофиза.
Третичные эндокринопатии – обусловлены повреждением гипоталамуса.
Все эндокринопатии могут проявляться по гипофункциональному и гиперфункциональному типу.
Причиной первичных гипофункциональных эндокринных нарушений является повреждение периферической железы, вместе с тем нередко наряду с дефицитом периферического гормона наблюдается повышение уровня тропного гормона аденогипофиза (по принципу обратной связи). Например, при болезни Аддисона дефицит кортизола сочетается избытком АКТГ. Только периферическим характером отмечаются гипофункциональные синдромы при повреждениях островков поджелудочной железы или околощитовидных желез.
Причиной первичных гиперфункциональных эндокринопатий обычно является опухоль, отличающаяся высокой секреторной активностью, выходящая из "подчинения регулирования обратной связью". Характерным в этом случае является понижение в крови соответствующего аденогипофизарного тропина.
Вторичные гипофункциональные нарушения редко изолированы (дефицит ТТГ - центральный гипотиреоз, дефицит АКТГ - центральный гипокортицизм), чаще страдают все гипофизарные функции с возникновением картины пангипопитуитаризма. Дифференциально диагностическим признаком по сравнению с первичными гипофункциями является сохранившаяся реакция на соответствующий гипофизарный тропин (правда, длительная нехватка аденогипофизарной стимуляции приводит зачастую к атрофии периферической железы). Уровень эндогенных тропинов в крови низкий.
Вторичные гиперфункциональные эндокринопатии обычно вызваны аденомой аденогипофиза и им присущ характер изолированного гиперпитуитаризма (чаше всего базофильная аденома - болезнь Иценко-Кушинга, несколько реже ацидофильная аденома - гигантизм или акромегалия).
Третичные эндокринопатии чаще всего проявляются в области гонадотропинов (эмоциональная аменорея, случаи преждевременного полового созревания).
Рис. Комплексная схема сложной обратной связи с приведением модулирующих факторов влияния вышестоящих нервных центров.
Эндокринная дисфункция - комбинации гипо- и гиперфункциональных нарушений железы или изменение качества ее секреции.
Типичным примером может служить адреногенитальный синдром, когда первичное понижение синтеза и секреции кортизола по принципу обратной связи увеличивает секрецию АКТГ, который способен повышать продукцию надпочечниковых половых гормонов. Следовательно, имеет место в одной железе гипофункция (глюкокортикоидная) и гиперфункция (половых гормонов).
87. – Патофизиологическая характеристика основных вариантов гиперпитуитаризма.
Общие механизмы эндокринных нарушений можно разделить по местоположению первичного повреждения на две большие группы, железистые и внежелезистые эндокринопатии.
Железистые эндокринопатии представляют собой нарушения самих эндокринных желез, когда первичное повреждение сводится к одной из желез внутренней секреции; они занимают самую большую долю из общей численности эндокринопатий.
В соответствии с субординационной структурой внутренней секреции большинства гормонов патогенетически различают следующие виды железистых эндокринопатий:
Первичные эндокринопатии – нарушение гормонообразования является следствием повреждения периферической железы.
Вторичные эндокринопатии – обусловлены повреждением гипофиза.
Третичные эндокринопатии – обусловлены повреждением гипоталамуса.
Все эндокринопатии могут проявляться по гипофункциональному и гиперфункциональному типу.
Причиной первичных гипофункциональных эндокринных нарушений является повреждение периферической железы, вместе с тем нередко наряду с дефицитом периферического гормона наблюдается повышение уровня тропного гормона аденогипофиза (по принципу обратной связи). Например, при болезни Аддисона дефицит кортизола сочетается избытком АКТГ. Только периферическим характером отмечаются гипофункциональные синдромы при повреждениях островков поджелудочной железы или околощитовидных желез.
Причиной первичных гиперфункциональных эндокринопатий обычно является опухоль, отличающаяся высокой секреторной активностью, выходящая из "подчинения регулирования обратной связью". Характерным в этом случае является понижение в крови соответствующего аденогипофизарного тропина.
Вторичные гипофункциональные нарушения редко изолированы (дефицит ТТГ - центральный гипотиреоз, дефицит АКТГ - центральный гипокортицизм), чаще страдают все гипофизарные функции с возникновением картины пангипопитуитаризма. Дифференциально диагностическим признаком по сравнению с первичными гипофункциями является сохранившаяся реакция на соответствующий гипофизарный тропин (правда, длительная нехватка аденогипофизарной стимуляции приводит зачастую к атрофии периферической железы). Уровень эндогенных тропинов в крови низкий.
Вторичные гиперфункциональные эндокринопатии обычно вызваны аденомой аденогипофиза и им присущ характер изолированного гиперпитуитаризма (чаше всего базофильная аденома - болезнь Иценко-Кушинга, несколько реже ацидофильная аденома - гигантизм или акромегалия).
Третичные эндокринопатии чаще всего проявляются в области гонадотропинов (эмоциональная аменорея, случаи преждевременного полового созревания).
Рис. Комплексная схема сложной обратной связи с приведением модулирующих факторов влияния вышестоящих нервных центров.
Эндокринная дисфункция - комбинации гипо- и гиперфункциональных нарушений железы или изменение качества ее секреции.
Типичным примером может служить адреногенитальный синдром, когда первичное понижение синтеза и секреции кортизола по принципу обратной связи увеличивает секрецию АКТГ, который способен повышать продукцию надпочечниковых половых гормонов. Следовательно, имеет место в одной железе гипофункция (глюкокортикоидная) и гиперфункция (половых гормонов).
88. - Патофизиологическая характеристика недостаточности пучковой зоны коры надпочечников. Этиология и патогенез болезни Аддисона.
Первичный гипокортицизм (периферический):
аутоиммунное поражение надпочечников (нередко сочетающееся с аутоиммунным поражением других эндокринных желез - щитовидной, околощитовидных, поджелудочной железы, яичников, а также кожи);
бактериальная и грибковая инфекции (туберкулез, бластомикоз, гистоплазмоз, менингококковая инфекция, сепсис различной этиологии);
удаление надпочечников по поводу болезни Иценко-Кушинга и других заболеваний;
амилоидоз;
гемохроматоз;
как осложнение при использовании различных лекарственных средств (антикоагулянты, блокаторы стероидогенеза в надпочечниках - аминоглютатемид, хлодитан, кетоконазол; барбитураты, спиронолактон).
Вторичный и третичный гипокортицизмы (центральные):
изолированный дефицит АКТГ (встречается крайне редко);
опухоли гипофиза и гипоталамуса (аденомы, краниофарингиомы, герминомы и пр.).
сосудистые заболевания (аневризма сонной артерии, кровоизлияние в гипофиз и в аденому гипофиза);
гранулематозные процессы в области гипофиза или гипоталамуса (саркоидоз, сифилис, гранулематозный гипофизит, аутоиммунный гипофизит);
деструктивно-травматические причины (операции, облучение гипофиза и гипоталамуса, удаление опухоли гипофиза и др.);
при резком прекращении приема глюкокортикоидов в ходе длительного курса фармакотерапии ("синдром отмены"); такая острая форма (адреналовый криз) проявляется тяжелыми явлениями гипотензии, электролитными нарушениями, адинамией.
Гипофизарный гипокортицизм - нередко составная часть пангипопитуитаризма, обычно менее выразителен, чем первичный, периферический гипокортицизм, вызванный патологией самой коры надпочечников.
Хроническая форма гипокортицизма (периферический вариант - болезнь Аддисона) проявляется слабостью, гипотензией, потерями натрия и гиперкалиемией.
Мышечная слабость связана с нарушением электролитного баланса (дефицит альдостерона) и гипогликемией (дефицит глюкокортикоидов), а также уменьшением мышечной массы (вследствие дефицита андрогенов). Артериальная гипотензия связана с гипонатриемией, выпадением пермиссивного эффекта глюкокортикоидов и вследствие этого снижения реактивных свойств сосудистой стенки к прессорным влияниям. Гипотензия может усугубиться ослаблением сократительной функции сердца.
Потеря натрия сопровождается полиурией, гипогидратацией, сгущением крови. Наряду с артериальной гипотензией ухудшение реологических свойств крови приводит к уменьшению клубочкового кровотока, эффективного фильтрационного давления и общего объема фильтрации. Отсюда наряду с полиурией может возникать недостаточность выделительной функции почек. Пищеварительные расстройства связывают с недостаточной секрецией пищеварительных соков и интенсивным выделением слизистой оболочкой кишечника ионов натрия (дефицит альдостерона), что приводит к профузным поносам, а также способствует гипогидратации.
Отличие центрального от периферического гипокортицизма заключается в отсутствии пигментации (избыток АКТГ стимулирует меланоциты) и сохраняющейся реакцией надпочечников на АКТГ.
89. - Патофизиологическая характеристика гиперкортизолизма. Дифференциальная диагностика болезни и синдрома Иценко-Кушинга.
Гиперкортицизм
Патологический гиперкортицизм
Эндогенный гиперкортицизм
Болезнь Иценко-Кушинга (центральный гиперкортицизм) – сложное нейро-эндокринное заболевание гипоталамо-гипофизарного генеза с последующим вовлечением надпочечников и формированием синдрома тотального гиперкортицизма и связанного с ним нарушения всех видов обмена веществ: белкового, жирового, углеводного и минерального. Патогенез проистекает из повышенной секреции АКТГ, этиология - притупление рецепторов гипоталамуса к глюкокортикоидам и базофильная аденома гипофиза.
Синдром Иценко-Кушинга (периферический гиперкортицизм) - эндогенный гиперкортицизм, развившийся вследствие первичного поражения коры надпочечника (доброкачественная или злокачественная опухоль - кортикостерома или двухсторонняя мелкоузелковая дисплазия коры надпочечников) или опухоли АПУД-системы.
АКТГ-эктопированный синдром - опухоли бронхов, поджелудочной железы, тимуса, печени, яичников, секретирующие АКТГ или кортикотропин-рилизинг фактор.