

13.Энзимопатии: понятие, классификация, молекулярные причины возникновения и механизмы развития, последствия, биохимическая диагностика.
I. Энзимопатология
Энзимопатология – это наука, которая изучает энзимопатии.
Энзимопатии – это группа заболеваний, которые вызваны различными дефектами ферментов. Энзимопатий делятся на: наследственные (первичные) и приобретенные (вторичные).
1. Наследственные энзимопатии
Наследственные энзимопатии – это заболевания, вызванные наследственными нарушениями биосинтеза ферментов или их структуры и функции.
В норме:

Полное или частичное нарушения биосинтеза ферментов вызывают дефекты генов регуляторных белков, которые контролируют синтез ферментов:
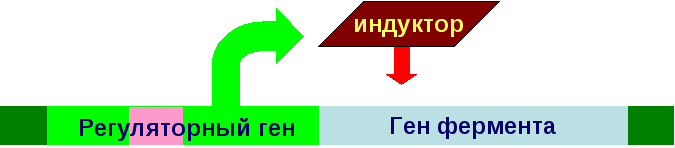
Нарушение структуры и функции ферментов вызывают дефекты генов этих ферментов:

У образовавшегося фермента наблюдаются структурные изменения, которые проявляются в изменении его каталитической активности (как правило, она исчезает), чувствительности к активаторам и ингибиторам, сродству к субстратам, оптимумам рН, температуры. В связи с этим изучением констант фермента является решающим в постановке диагноза врожденных энзимопатий.
Наследственные энзимопатии по типу нарушений метаболизма делят на:
нарушения обмена аминокислот: фенилкетонурия, альбинизм, алкаптонурия и др.;
нарушения углеводного обмена: галактоземия, наследственная непереносимость фруктозы, гликогенозы;
нарушения липидного обмена: липидозы;
нарушения обмена нуклеиновых оснований: подагры, синдрома Леш-Нихана и др.;
нарушение обмена в соединительной ткани: мукополисахаридозы, хондродистрофия и др.;
дефекты ферментов в ЖКТ: муковисцидоз, целиакия, непереносимость лактозы и др.
нарушения обмена стероидов и т.д.
В норме метаболический путь протекает следующим образом:
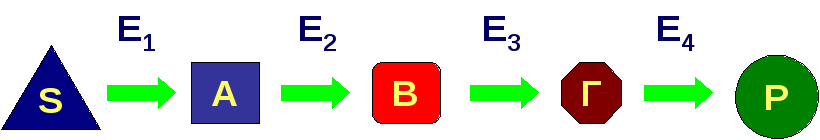
Из-за дефекта в метаболическом пути (цикле, шунте) одного из ферментов в организме происходит накопление промежуточных продуктов (часто токсичных в высоких концентрациях) и дефицит жизненно необходимых конечных продуктов, что приводит к клиническим проявлениям:

Пример: фенилпировиноградная олигофрения – наследственное заболевание, приводящее в раннем детстве к гибели ребенка или к развитию у него тяжелой умственной отсталости.
Причиной заболевания является отсутствие в печени фермента фен-4-монооксигеназы, которая обеспечивает превращение незаменимой аминокислоты Фен в Тир:
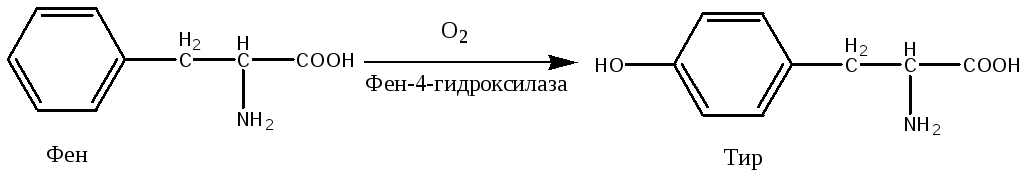
Эта реакция необходима для катаболизма Фен, т.е. удаления его излишков. При отсутствии фен-4-монооксигеназы в организме происходит накопление Фен и превращение его в различные производные: фенилпировиноградную, фенилмолочную и фенилуксусную кислоты.
Фен и его производные в высоких концентрациях токсичны, накапливаясь в тканях, они оказывают на них повреждающее действие. Самой чувствительной к Фен и его производным оказывается нервная ткань детей, она поражается в первую очередь.
Диагноз фенилкетонурия ставят на основании обнаружения Фен в крови или фенилпировиноградной кислоты на пеленках детей. Лечение в основном сводится к исключению из питания ребенка Фен. Для такого ребенка Тир оказывается незаменимой аминокислотой.
Другое тяжелое наследственное заболевание – галактеземия (непереносимость молочного сахара), связано с отсутствием синтеза в печени ферментов, катализирующих превращение галактозы в глюкозу. В результате в раннем возврате происходит накопление в тканях галактозы, приводящее к развитию катаракты, поражению печени, мозга, нередко вызывающее гибель ребенка. Лечение в данном случае сводиться к исключению из диеты молочного сахара.