

3.4. Биосинтез креатина и его последующие превращения.
3.4.1. Синтез креатина в тканях человека протекает в две стадии. На первой стадии в почках образуется гуанидинацетат:

На второй стадии в печени происходит реакция трансметилирования:

3.4.2. Синтезированный в печени креатин поступает в кровь и доставляется в мышцы. Там он взаимодействует с АТФ, в результате чего образуется макроэргическое соединение креатинфосфат. Эта реакция легко обратима.

В состоянии покоя мышцы накапливают креатинфосфат (его содержание в неработающей мышце в 3-8 раз выше, чем содержание АТФ). При переходе к мышечной работе изменяется направление реакции и образуется АТФ, необходимый для мышечного сокращения.
Образование АТФ при участии креатинфосфата – наиболее быстрый путь генерации АТФ. Запас креатинфосфата обеспечивает интенсивную работу мышц в течение 2 – 5 секунд. За это время человек успевает пробежать 15 – 50 метров. Тем временем включаются другие механизмы образования АТФ: мобилизация мышечного гликогена, окисление субстратов, поступающих из печени и жировой ткани.
Концентрация креатина в крови здоровых взрослых людей составляет приблизительно 50 мкмоль/л; в моче он практически отсутствует. Появление креатина в моче не всегда является симптомом заболевания. Так, у маленьких детей и подростков моча всегда содержит креатин (физиологическая креатинурия). При заболеваниях мышц, когда нарушается образование креатинфосфата, увеличивается содержание креатина в крови и возрастает его экскреция с мочой.
3.4.3. В результате неферментативного дефосфорилирования креатинфосфата образуется креатинин – ангидрид креатина.

Креатинин – один из конечных продуктов азотистого обмена в организме, он выводится с мочой. Суточное выделение креатинина у здорового человека пропорционально его мышечной массе. Креатинин не реабсорбируется в почечных канальцах, поэтому его суточная экскреция является показателем фильтрационной функции почек. Содержание креатинина в крови снижается при заболеваниях мышц и увеличивается при нарушении функции почек. Выделение креатинина с мочой снижается в обоих случаях.
3.5. Обмен фенилаланина и тирозина.
3.5.1. Обмен фенилаланина и тирозина в тканях человека можно представить в следующем виде (см. рисунок 3.2).
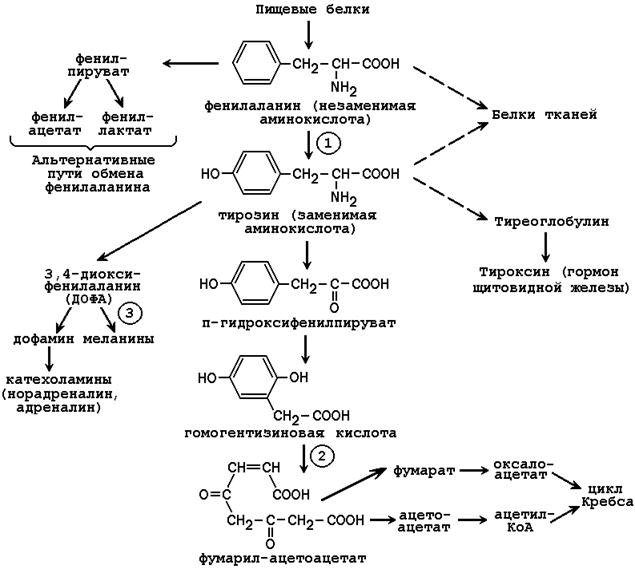
Рисунок 3.2. Пути обмена фенилаланина и тирозина в тканях (цифрами обозначены наиболее часто встречающиеся дефекты ферментов; далее приводится характеристика этих нарушений).
3.5.2. Известен ряд врождённых нарушений обмена фенилаланина и тирозина.
Фенилкетонурия – врождённое нарушение процесса гидроксилирования фенилаланина до тирозина. Заболевание чаще всего вызвано отсутствием или недостатком фермента фенилаланингидроксилазы (обозначен цифрой 1 на рисунке 3.2), реже - нарушением образования тетрагидробиоптерина.
Ранними симптомами фенилкетонурии являются повышенная возбудимость и двигательная активность, рвота и трудности вскармливания, с 3 – 5-го месяца нарушается интеллектуальное развитие, исчезает реакция на окружающее. Со временем у детей появляются судороги. Волосы и глаза обычно менее пигментированы, чем у других членов семьи. При отсутствии лечения продолжительность жизни больных составляет 20 - 30 лет.
Биохимическая основа фенилкетонурии – накопление фенилаланина в организме. Высокая концентрация аминокислоты стимулирует выработку фермента, превращающего фенилаланин в фенилпируват (в норме этот фермент малоактивен). Путём восстановления фенилпируват переходит в фениллактат, а путём декарбоксилирования – в фенилацетат. Эти продукты наряду с фенилаланином в существенных количествах обнаруживаются в моче больных.
В настоящее время имеются достоверные свидетельства того, что за токсическое повреждение мозга ответственны главным образом высокие концентрации фенилаланина. Повышенное содержание фенилаланина тормозит транспорт тирозина и других аминокислот через биологические мембраны. Это приводит к ограничению синтеза белка в клетках мозга и нарушению синтеза нейромедиаторов.
Раннюю диагностику заболевания нельзя провести исходя только из клинической симптоматики. Диагноз ставится биохимически путём скрининга всех новорождённых. Лечение больных фенилкетонурией основано на ограничении поступления фенилаланина в организм и снижения концентрации этой аминокислоты в плазме. С этой целью используются искусственные питательные смеси, в которых фенилаланин отсутствует (например, берлофен).
Алкаптонурия – врожденное нарушение обмена фенилаланина, вызванное отсутствием фермента оксидазы гомогентизиновой кислоты (цифра 2 на рисунке 3.2). Это приводит к нарушению образования малеилацетоацетата, расщепляющегося далее до фумарата и ацетоацетата. В раннем детском возрасте единственным проявлением дефицита фермента является изменение окраски мочи. Гомогентизиновая кислота секретируется в просвет канальцев и в значительном количестве выводится с мочой. На воздухе она окисляется, а затем полимеризуется в окрашенное соединение, которое окрашивает пелёнки в чёрный цвет. Экскреция гомогентизиновой кислоты зависит от содержания фенилаланина и тирозина в пище.
Следствием накопления гомогентизиновой кислоты в организме является охроноз - шиферно-голубой оттенок ушного и носового хрящей, вызванный накоплением в них пигмента. Развитие охроноза можно предотвратить, если с раннего возраста ограничивать поступление с пищей фенилаланина и тирозина.
Альбинизм развивается при отсутствии в пигментных клетках фермента тирозиназы (обозначена цифрой 3 на рисунке 3.2), которая участвует в образовании меланина. В результате волосы, кожа и глаза больного лишены этого пигмента. При альбинизме наблюдается повышение чувствительности к солнечным лучам и некоторое нарушения зрения.
Контрольные вопросы:
Перечислите источники и пути включения азота в состав аминокислот.
Представьте в виде схемы образование углеродного скелета аланина из глюкозы. Укажите источники азота для синтеза аланина и напишите соответствующие реакции.
Представьте в виде схемы образование углеродного скелета аспартата из глюкозы. Укажите источники азота для синтеза аспартата и напишите соответствующие реакции.
Представьте в виде схемы синтез углеродного скелета глутамата из глюкозы. Укажите источники азота для синтеза глутамата и напишите соответствующие реакции.
Напишите реакции синтеза тирозина в организме с участием незаменимой аминокислоты, укажите название фермента.
Напишите реакции синтеза цистеина в организме с участием незаменимой аминокислоты, укажите названия ферментов.
Назовите аминокислоты – источники одноуглеродных радикалов и напишите формулу кофермента, который участвует в переносе этих радикалов.
Перечислите одноуглеродные производные ТГФК и пути использования их в реакциях биосинтеза.
Напишите формулу S-аденозил-метионина и перечислите вещества, в синтезе которых используется метильная группа.
Напишите реакцию образования метионина из гомоцистеина, укажите источник метильной группы и биологическую роль этой реакции.
Напишите реакции синтеза креатина и укажите их локализацию и катализирующие их ферменты.
Напишите реакцию образования креатинфосфата из креатина. Назовите фермент, катализирующий эту реакцию и укажите биологическую роль креатинфосфата.
Напишите реакцию образования креатинина. Укажите факторы, влияющие на суточную экскрецию креатинина в норме и при патологии.
Перечислите биологически активные вещества, синтезируемые из фенилаланина и тирозина, укажите их роль в организме.
Представьте в виде схемы образование гомогентизиновой кислоты из фенилаланина. При каком заболевании гомогентизиновая кислота обнаруживается в моче?
Представьте в виде схемы включение углеродного скелета фенилаланина в глюкозу и кетоновые тела.
Представьте в виде схемы включение углеродного скелета тирозина в цикл Кребса.
Какие патологические компоненты обнаруживаются в моче при врождённых нарушениях обмена фенилаланина и тирозина?