

Диагностика
Для диагностики ПМФ необходимо выполнить клинический анализ крови, рентгенографию или МРТ костей (неоднородное повышение плотности), МРТ (КТ, УЗИ) селезенки и печени, аспирацию и биопсию КМ, цитогенетическое исследование КМ и/или периферической крови (FISH для выявления цитогенети-ческих аномалий), ПЦР лейкоцитов периферической крови (или КМ) для выявления мутации JAK2V617F, (а также для исключения bcr/abl). Классическими критериями диагноза ПМФ ранее являлись сплено-мегалия, коллагеновый миелофиброз и лейкоэритро-бластическая картина периферической крови. В соответствии с критериями ВОЗ (2008), современный диагноз ПМФ базируется на оценке клинических, морфологических, цитогенетических и молекулярных данных (табл. 22.1).
На основе Европейской консенсусной классификации по градации миелофиброза (2005) выделяют три степени миелофиброза (табл. 22.2).
Необходимо также разграничивать миелофиброз, связанный с прогрессированием ИП и ЭТ (табл. 22.3.).
Таблица 22.1. Критерии диагностики первичного миелофиброза (Всемирная организация здравоохранения, 2008)
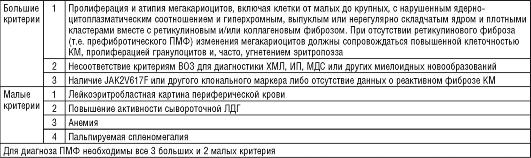
Таблица 22.2. Европейская консенсусная классификация по градации миелофиброза (Thiele J. et al., 2005)

Таблица 22.3. Критерии диагностики постполицитемического/посттромбоцитемического миелофиброза (International Working Group for Myelofibrosis Research and Treatment criteria, 2008)

*Необходимо ≥2 критерия.
При дифференциальном диагнозе следует исключить заболевания, которые могут быть причиной развития миелофиброза. Некоторыми авторами отмечено снижение выраженности клинических симптомов у больных ПМФ за период в 15 лет. Так, у пациентов, диагностированных до 1987 г., более часто определяли конституциональные симптомы (лихорадку, ночную потливость, снижение массы тела), более высокую частоту спленомегалии и гепатомегалии и более часто фазу остеосклероза при диагностике, чем у тех, у кого диагноз был установлен в 1990-х гг. Однако при этом не найдено значимой разницы в прогностических факторах и выживаемости между этими группами больных.
Одним из основных методов диагностики ПМФ остается трепанобиопсия с последующим гистологическим и/или гистоморфометрическим исследованием КМ. В связи с клинико-гистологическими различиями начальной префибротической фазы ПМФ и продвинутых фаз с развитием коллагенового фиброза КМ используется термин, отражающий гистологические особенности ранней (пролиферативной) фазы ПМФ - эссенциальная мегакариоцитарно-гранулоцитарная метаплазия. При эссенциальной мегакариоцитарно-гранулоцитарной метаплазии наблюдается неопластическая пролиферация нарушенного мегакариоци-тарного и гранулоцитарного ростков. Миелоидная метаплазия может встречаться уже и на этой фазе. Гистопатологически в КМ, как при эссенциальной мегакариоцитарно-гранулоцитарной метаплазии, так и классическом ПМФ, доминируют атипичные, увеличенные и незрелые мегакариоциты с облаковид-ными незрелыми диспластическими ядрами, которые не наблюдаются при ЭТ и ИП. Особенности мегака-риопоэза могут быть отличительным диагностическим признаком пролиферативной (префибротической) фазы ПМФ, разграничивающим ПМФ и другие кМПЗ.
Миелофиброз при ПМФ, как и при всех кМПЗ, является генерализованным прогрессирующим с разной скоростью процессом с неоднородной по степени выраженностью в разных областях КМ - в подвздошных костях, позвонках и грудине. Эволюция фиброза КМ в значительной степени связана с преобладанием больших атипичных, возможно, длительно живущих и гиперплоидных мегакариоцитов, а не с увеличением клеток-предшественников. В то же время при миело-идной метаплазии селезенки, связанной с ПМФ, мега-кариоцитопоэз селезенки имеет значительные отли-
чия: мегакариоциты уменьшенных размеров, нарушено их ядерно-цитоплазматическое соотношение, определяется относительное увеличение частоты промегака-риобластов; в целом, экстрамедуллярный мегакариоци-топоэз отличается более высокой степенью незрелости, чем костномозговой мегакариоцитопоэз при ПМФ.
Миелофиброз сопровождает развитие всех миело-пролиферативных заболеваний и проявляется в разной степени выраженности при разных заболеваниях, а данные относительно его прогрессирования остаются весьма гетерогенными. В работе M. Adamkov и соавт. (1998) при первичной диагностике на основе гистологического полуколичественного измерения импрегни-рованных ретикулиновых волокон миелофиброз выявлен более чем у 94,4% больных ПМФ, у 27,3% больных ИП, у 21% больных ЭТ, а также у 48% пациентов с ХМЛ. При повторных биопсиях наиболее частым было прогрессирование миелофиброза при ПМФ.
Миелофиброз КМ может возникать не только при ПМФ, но так же как исход при других кМПЗ - ИП, ЭТ. В то же время, поскольку фиброз КМ является неопухолевой реакцией стромальных клеток КМ, он может развиваться и при других, не связанных с кМПЗ заболеваниях. Вторичный фиброз КМ встречается и при некоторых других онкогематологиче-ских заболеваниях: ХМЛ, волосатоклеточном лейкозе, реже - при ОЛ, остром миелофиброзе - редком злокачественном заболевании системы крови с плохим прогнозом; при МДС («синдром перекреста»), злокачественных лимфомах. Фиброзирование КМ отмечается и при солидных опухолях с метастазами в КМ (рак предстательной железы, молочной железы, легких). Нередко фиброз КМ развивается при диффузных заболеваниях соединительной ткани (СКВ, системной склеродермии) - как аутоиммунный фиброз КМ, а также изредка как сосуществование двух заболеваний - тяжелого фиброза КМ и СКВ. Описано несколько случаев ассоциации фиброза КМ с висцеральным лейшманиозом, миелоидной метаплазией при туберкулезе, недостаточностью витамина D при рахите.