

Teplovue effektu i napravlenie himicheskih processov
.pdf11
вероятность и энтропия вещества. Энтропия растёт не только при нагревании, но и при эндотермических фазовых переходах: плавление, испарение и др. Энтропия резко возрастает при переходе вещества из жидкого состояния в газообразное.
Например, Sо298 н2о(ж) = 70,08, Sо298 н2о(г) = 188, 72 Дж/(моль К). Энтропия увеличивается при превращении вещества из кристаллического в аморфное. Чем прочнее вещество, тем упорядоченее его структура и тем меньше значение его абсолютной энтропии. Например, графит и алмаз являются модификациями углерода: графит – хрупкий, а алмаз – один из самых твердых кристаллов:
Sо298 графит = 5,740, Sо298 алмаз = 2,368 Дж/(моль К). Увеличение числа атомов в молекуле и усложнение молекул приводит к увеличению энтропии. Например, Sо298 (О)=160,9, Sо298 (О2 (г)) = 205,04, Sо298 (О3 (г)) = 238,8 Дж/(моль К).
Изменение энтропии в химической реакции определяется разностью сумм энтропий продуктов и энтропий исходных веществ.
Например, для реакции общего типа
а А + b В + ... → с С + d D + …
изменение энтропии, определяется выражением
Sо=[c Sо(C) + d Sо(D) + …] – [a Sо(A) + b Sо(B) + …],
где S – абсолютные значения энтропии всех веществ, Дж/(моль К) или
кал /(моль К):
Sо = ΣSопрод. – ΣSоисх.
Об изменении энтропии в химической реакции можно судить по изменению объёма системы в ходе реакции. Например, в реакции
? С(графит) + ? СО2 (г) = СО(г), Sо298 = 87,8 Дж/К
наблюдается увеличение объёма (ΔV>0); следовательно, энтальпия возрастает (ΔS>0). В случае же реакции образования аммиака из водорода и азота
3/2 Н2 (г) + ? N2 (г) = NН3 (г), |
Sо298 = – 103,1 Дж/К |
наоборот, объём системы уменьшается (ΔV<0); следовательно, энтропия |
|
уменьшается (ΔS<0) |
|
Если же реакция протекает между твердыми веществами, например, |
|
Al(к) + Sb(к) = AlSb(к), |
Sо298 = –5,1 Дж/К, |
то изменения объёма системы и её энтропии практически не происходит. То же относится и к процессам, в которых количество (моль) газообразных веществ не изменяется, например:
С(графит) + О2 (г) = СО2 (к), Sо298 = 2,9 Дж/К
12
5. ЭНЕРГИЯ ГИББСА
Самостоятельно, т.е. без затрат работы извне, система может переходить только из менее устойчивого состояния в более устойчивое. В химических процессах одновременно действуют две тенденции: стремление частиц объединиться за счёт прочных связей в более сложные, что уменьшает энтальпию системы, и стремление частиц разъединиться, что увеличивает энтропию. Иными словами, проявляется действие двух прямо противоположных факторов – энтальпийного ( Н) и энтропийного (T S). Суммарный эффект этих двух противоположных тенденций в процессах, протекающих при постоянных Т и р, отражает изменение энергии Гиббса G или изобарно–изотермического потенциала.
Энергия Гиббса G – величина, названная так в честь американского учёного Дж. Уилларда Гиббса (1839–1903), одного из основоположников химической термодинамики.
Согласно Н = G + T S теплота T S идет на создание беспорядка (бесполезно рассеивается в окружающую среду) и потому не может быть использована для совершения работы; её часто называют связанной энергией. Теплота G* может быть использована для совершения работы, и поэтому энергию Гиббса часто называют также свободной энергией:
G = Н – T S
Характер изменения энергии Гиббса позволяет судить о принципиальной возможности или невозможности осуществления процесса.
Условием принципиальной возможности процесса является неравенство G<0.
Иными словами, самопроизвольно протекают реакции, если энергия Гиббса в исходном состоянии системы больше, чем в конечном. Увеличение энергии Гиббса
G>0
свидетельствует о невозможности самопроизвольного осуществления процесса в данных условиях. Если же
G=0,
система находится в состоянии химического равновесия.
Энтальпийный и энтропийный факторы и направление процесса
В соответствии с уравнением
G = Н – T S
самопроизвольному протеканию процесса способствует уменьшение энтальпии и увеличение энтропии системы, т.е. когда Н<0 и S>0.
13
При других сочетаниях характера изменений Н и S возможность процесса определяет либо энтальпийный, либо энтропийный фактор.
Рассмотрим две следующие реакции:
Но298 |
CaO (к) + СО2 (г) = CaCO3 (к) |
= –178,0 кДж, Sо298 = –160,48 Дж/К |
|
|
Gо298 = –130,2 кДж |
Но298 |
CaCO3 (к) = CaO (к) + СО2 (г) |
= 178,0 кДж, Sо298 = 160,48 Дж/К |
Gо298 = –62,7 кДж
Первая реакция экзотермическая, протекает с уменьшением объёма. Возможность этой реакции (ΔG<0) определяется действием энтальпийного фактора, который перекрывает противодействие энтропийного фактора (по абсолютному значению |ΔН| >|T S|).
Вторая реакция эндотермическая, протекает с увеличением объёма. Возможность этой реакции (ΔG<0), наоборот, определяется энтропийным фактором. При высокой температуре энтропийный фактор перекрывает энтальпийный фактор (т.е. |ΔН| < |T S|) и реакция протекает самопроизвольно.
Влияние температуры на направление реакции |
|
|
|
|
Согласно уравнению G = |
Н – T S влияние температуры |
на |
G |
|
определяется знаком и величиной |
S. На рис. 2 показана зависимость |
G ряда |
||
реакций от температуры. Если пренебречь влиянием Т на значения |
Н и |
S, то |
||
приведённая зависимость G =?(T) |
|
|
S. На |
|
является уравнением прямой, наклон которой определяется знаком |
||||
рис. 2 при S>0 прямая идет вниз, при |
S<0 |
|||
– вверх. Для реакции |
|
|
|
|
|
2C (графит) + O2 (г) = 2СО(г), S>0, |
|
|
|
|
протекающей с увеличением энтропии, |
|||
повышение температуры приводит к |
G. |
|||
увеличению отрицательного значения |
||||
Высокотемпературный режим |
|
|
|
|
благоприятствует протеканию процесса. Для |
||||
реакции 2Hg (ж) + O2 (г) = 2HgO (к), |
S<0, |
|
||
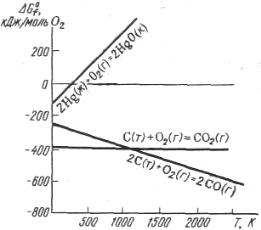
Рис. 2. Зависимость G ряда оксидов от температуры
14
протекающей с уменьшением энтропии, с повышением температуры отрицательное значение G уменьшается. Следовательно, в этом случае высокотемпературный режим препятствует протеканию процесса. При соответствующей температуре G приобретает положительное значение, и тогда реакция должна протекать в обратном направлении. Как видно на рис. 2, изменение знака G для этой реакции достигается при 500 К. Выше этой температуры реакция протекает в обратном направлении:
2HgO (к) = 2Hg (ж) + O2 (г), S>0
Таким образом, при низкотемпературном режиме (до 500 К) ртуть окисляется кислородом, в то время как при высокотемпературном режиме (выше 500 К) оксид ртути распадается с выделением кислорода. Эти процессы можно использовать для получения кислорода в лаборатории.
Если же при протекании процесса энтропия системы не изменяется, то значение G от температуры практически не зависит. Так, для реакции
C (графит) + O2 (г) = СО2 (г), |
S≈0 |
зависимость G =?(T) на рис.2 изображена прямой, практически параллельной |
|
оси абсцисс. |
энтальпии (ΔН < 0) и |
Процессы, протекающие с уменьшением |
|
увеличением энтропии (ΔS > 0), практически необратимы. В этом случае G всегда будет иметь отрицательное значение, какую бы температуру ни приняли.
Влияние температуры на направление химических реакций
Н |
S |
G |
Направление реакции |
Примеры реакций |
|
|
|
|
|
Н<0 |
S>0 |
G<0 |
Прямая реакция |
2C (графит) + O2 (г) = 2СО(г) |
|
|
|
может быть |
|
|
|
|
самопроизвольной при |
|
|
|
|
любых температурах |
|
Н>0 |
S<0 |
G>0 |
Прямая реакция не |
2СО(г) = 2C (графит) + O2 (г) |
|
|
|
может идти |
|
|
|
|
самопроизвольно при |
|
|
|
|
любых температурах |
|
Н<0 |
S<0 |
G<0 |
Самопроизвольно |
CaO (к) + СО2 (г) = CaCO3 (к) |
|
|
G>0 |
может идти прямая |
|
|
|
|
реакция при низких |
|
|
|
|
температурах и |
|
|
|
|
обратная реакция при |
|
|
|
|
высоких температурах |
|
Н>0 |
S>0 |
G>0 |
Самопроизвольно |
CH4 + 2H2O(г) = СО2 (г) + 4H2(г) |
|
|
G<0 |
может протекать |
|
|
|
|
прямая реакция при |
|
|
|
|
высоких температурах |
|
|
|
|
и обратная реакция при |
|
|
|
|
низких температурах |
|
15
Есть случаи, когда реакция термодинамически разрешена, а
самопроизвольно не идёт. Например: |
G = – 474,38 кДж |
2H2(г) + O2 (г) = 2H2O(ж), |
В обычных условиях эта реакция практически не идёт. Но стоит внести в смесь подходящий катализатор (мелкодисперсную платину) или просто поднести горящую спичку, реакция произойдёт со взрывом: это гремучий газ.
Если реакция термодинамически не разрешена, условия подобрать невозможно.
6. КОНСТАНТА РАВНОВЕСИЯ И ИЗОБАРНО–ИЗОТЕРМИЧЕСКИЙ ПОТЕНЦИАЛ РЕАКЦИИ
Константа химического равновесия зависит от природы реагентов и температуры. Она связана с изменением стандартной энергии Гиббса химической реакции Gо уравнением
Gо = –RT ln K, Gо298 (кДж) = –5,71 lg K298 |
(7) |
Приведённое уравнение позволяет по величине |
Gо вычислить К, а затем |
и равновесные концентрации (парциальные давления) реагентов. Большим отрицательным значениям Gо ( G<<0) отвечают большие значения К (К>>1), т.е. в равновесной смеси преобладают продукты взаимодействия. При больших положительных значениях Gо ( Gо>>0) в равновесной смеси преобладают
исходные вещества (К<<1). Если учесть, что |
G = Н – T S = –RT lnK, то |
|||
после некоторого преобразования получим |
|
|
||
|
H |
S |
(8) |
|
RT e |
R |
|||
K e |
|
|||
Из этого уравнения видно, что константа равновесия очень чувствительна к изменению температуры. Для эндотермических процессов повышение температуры отвечает увеличению константы равновесия, для экзотермических
– её уменьшению. От давления (если оно не очень велико) константа равновесия не зависит.
Зависимость константы равновесия от энтальпийного и энтропийного факторов свидетельствует о влиянии на нее природы реагентов.
В литературе применяются термины–синонимы: свободная энергия, свободная энтальпия, свободная энергия при постоянном давлении, потенциал Гиббса, функция Гиббса, энергия Гиббса, изобарный потенциал, изобарно– изотермический потенциал
16
7. ТЕРМОХИМИЧЕСКИЕ ИЗМЕРЕНИЯ И ВЫЧИСЛЕНИЯ
Измерение тепловых эффектов называется калориметрией. Методика и оборудование, применяемые в калориметрии, зависят от характера изучаемого процесса. Реакции горения обычно изучают при помощи так называемой калориметрической бомбы. Реакции в калориметрической бомбе протекают при постоянном объеме.
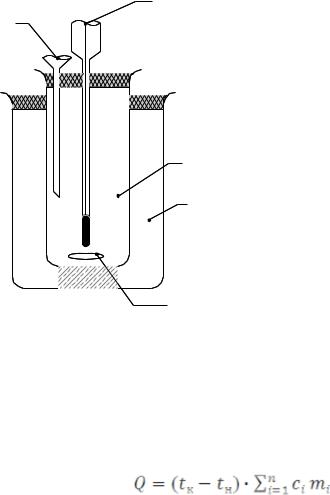
Большинство реакций осуществляется при постоянном давлении. Измерение изменения энтальпии этих реакций осуществляется в калориметрах различных конструкций, которые представляют собой по возможности лучше изолированный от теплообмена с внешней средой сосуд с мешалкой и термометром.
3 |
4 |
Упрощенный калориметр (рис.3) состоит из двух стаканов: наружного (1) и внутреннего, реакционного (2).
Реакционный стакан с магнитной мешалкой (5) закрывается крышкой с отверстиями: для термометра (3); для воронки
(4).
2
1
5
Рис. 3. Устройство калориметра
Количество теплоты, выделяющейся или поглощающейся в калориметре, определяется по формуле
, |
(9) |
где tк – конечная температура; tн– начальная температура;
сi – удельные теплоемкости калориметрического сосуда, калориметрической жидкости и исследуемого вещества;
mi – массы реакционного сосуда, жидкости и исследуемого вещества.
17
При использовании стеклянного реакционного сосуда теплоемкостью калориметрического сосуда можно пренебречь из-за его малой теплопроводности. Удельную теплоемкость растворов можно принять равной теплоемкости для воды: 4,2 кДж/кг град или 1 ккал/кг·град.
Тогда уравнение (10) примет следующий вид для стеклянного сосуда:
Q = (tк – tн)·4,2·m (кДж) или
(10)
Q = (tк – tн)·1·m (ккал),
где m –масса воды растворенного вещества.
(Масса воды или растворов находится как произведение объема жидкocти на ее плотность, т.е. m=ρ∙V. Плотность воды ρ равна 1 г/мл, плотность разбавленных растворов считать равной также 1 г/мл).
Пересчет теплового эффекта на 1 моль вещества, т.е. нахождение величины ∆H, производится по формуле
H |
Q |
, |
(11) |
|
n |
||||
|
|
|
где n – число молей вещества.
Все работы по определению изменения энтальпии химических процессов необходимо проводить в следующей последовательности.
1.Поместить в реакционный сосуд измеренный мензуркой объем воды или раствора и дождаться выравнивания температуры калориметра и окружающей среды. Для этого, выждав 3–5 мин, записывать показания термометра в течение трех минут с интервалом в 1 мин. Если температура практически не меняется, принять ее за величину tн.
2.Быстро внести через сухую воронку взвешенное на весах или измеренное цилиндром количество реагирующего вещества и, непрерывно перемешивая раствор мешалкой, производить замеры температуры через 0,5– 1 мин.
Результаты наблюдений записывать в виде таблицы:
Время отначала |
0 |
0,5 |
1,0 |
1,5 |
2,0 |
2,5 |
3,0 |
3,5 |
4,0 |
5,0 |
6,0 |
опыта, мин |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Температура, °С |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
3. Построить график изменения температуры во времени на миллиметровой бумаге, отложив на оси ординат температуру, а на оси абсцисс
– время в минутах. На рис.4 приведен примерный вид этого графика.
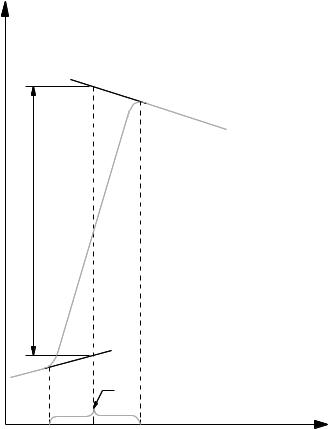
18
4. С помощью полученного графика найти изменение температуры в ходе опыта, ∆t = tк – tн. Для этого произвести экстраполяцию плавного линейного хода температуры конечного и исходного периодов времени (если температура
в начале |
опыта |
не менялась, |
то прямая этого участка зависимости будет |
|||||||
|
|
|
|
|
|
параллельна оси абсцисс). |
|
|||
|
|
|
|
|
|
|
Отрезок |
времени, |
за |
|
|
|
|
B |
|
|
который наблюдалось резкое |
||||
|
|
|
|
|
изменение |
|
температуры, |
|||
|
|
|
|
|
|
|
||||
, ?С |
|
|
|
|
M |
разделить |
пополам, |
из |
||
|
|
|
|
|
полученной |
|
точки |
|||
а |
|
|
|
|
|
|
||||
|
|
|
|
|
восстановить |
перпендикуляр |
||||
ер атур |
|
|
|
|
|
|||||
t |
|
|
|
|
до |
пересечения |
с |
|||
|
|
|
|
|
экстраполированными |
|||||
мп |
|
|
|
|
|
прямыми BM и NA. |
|
|||
те |
|
|
|
|
|
|
Величина |
отрезка |
||
|
|
|
|
|
|
перпендикуляра |
|
АВ, |
||
|
|
|
|
|
|
отсекаемого |
этими прямыми, |
|||
|
|
|
|
|
|
соответствует |
изменению |
|||
|
|
|
|
A |
|
температуры |
|
|
|
|
|
|
|
|
|
|
∆t = tк – tн. |
|
|
||
|
N |
|
|
интервалрезкогоизменениятемпературы |
|
|
||||
|
0 |
1.0 |
2.0 |
3.0 |
4.0 |
5.0 |
|
|
|
|
|
|
|
|
|
|
время,мин. |
|
|
|
|
Рис. 4. Зависимость температуры от времени
Используя описанные выше рекомендации, произведите измерение изменения температуры и вычислите значение энтальпии для некоторых процессов.
8.ЛАБОРАТОРНЫЕ РАБОТЫ
8.1.Определение энтальпии реакции нейтрализации
Реакции между растворами сильных кислот и сильных оснований, например:
NaOH + HCl→ NaCl+H2O,
KOH + ? H 2SO4→½ K2SO4 + H2O,
19
выражаются общим для них термохимическим уравнением
H+ + OH–→ H2O; .∆H= – 56,9 кДж/моль.
Теплота реакций нейтрализации слабых электролитов меньше 56,9Дж/моль. Значение теплоты реакции нейтрализации в этих случаях не является постоянным, т.к. оно зависит от природы слабой кислоты и слабого основания. Это объясняется тем, что экзотермическому процессу образования воды из ионов H+ и OH– в случае взаимодействия слабых электролитов предшествует эндотермический процесс диссоциации электролитов (кислот и оснований). Чем меньше количество тепла, выделяющееся в результате реакции нейтрализации слабого электролита, тем больше энергии поглощается при его диссоциации, тем слабее электролит. Таким образом, силу электролита можно сопоставить, сравнивая величину
∆Hнейтр. сл.эл–та – (–56,9) = .∆Hдисс. сл.эл–та
Чем больше эта величина, тем слабее электролит.
Выполнение работы
1. Получить у лаборанта растворы необходимых веществ:
а) NaOH и HCl;
б) NH4OH и HCl;
в) NaOH и CH3COOH;
г) NH4OH и CH3COOH;
д) KOH и HNO3.
2.Налить во внутренний стакан калориметра 40 мл раствора электролита с меньшей концентрацией (0,3–0,5 – молярный раствор). Измерить начальную температуру раствора (при работающей мешалке).
3.Быстро влить через воронку 10 мл электролита с большей концентрацией (1–2 –молярный раствор). Произвести измерение по методике, описанной в общих указаниях (гл. 7).
4.Рассчитать тепловые эффекты по формуле (10) и энтальпии реакции нейтрализации по формуле (11):
Н |
Q |
, где n – число молей электролита, взятого в недостатке. |
|
n |
|||
|
|
20
5. Оформить результаты работы и расчетов в виде следующей таблицы:
Система |
V, |
М, |
n |
MV |
|
∆t, |
Q, |
∆H, |
мл |
моль/л |
|
оС |
кДж |
кДж/моль |
|||
|
1000 |
|||||||
Кислота |
|
|
|
|
|
|
|
|
Основание |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6.Сделать вывод по результатам работы.
7.Составить молекулярные и сокращенные уравнения реакций взаимодействия указанных электролитов.
8.2.Определение энтальпии реакции присоединения кристаллизационной воды к безводной соли (энтальпия гидратации)
Энтальпию реакции присоединения кристаллизационной воды к безводной соли можно определить, зная изменение энтальпии при растворении безводной соли и изменение энтальпии при растворении кристаллогидрата этой соли.
В термохимии под энтальпией растворения понимают изменение энтальпии процесса растворения 1 моля вещества в большом количестве растворителя, т.е. в таком его количестве, когда дальнейшее разбавление раствора не влияет на величину энтальпии растворения (это так называемая дифференциальная энтальпия растворения). Энтальпия растворения веществ состоит в основном из двух слагаемых: энтальпии процесса перехода твердого или газообразного вещества в то состояние, в котором оно существует в растворе ∆Hр, и энтальпии процесса взаимодействия вещества с растворителем (энтальпия сольватации) ∆Hс. В зависимости от величины и знака этих двух слагаемых процесс растворения может быть эндотермическим или экзотермическим.
Для газов ∆Hр < 0 и величина ∆Hр представляет собой энтальпию конденсации при изменении объема газа до объема жидкого раствора.