


Highly Accurate Ab Initio Computation of Thermochemical Data |
11 |
4.2.Dependence on the AO Basis Set
In our discussion so far, we have used electronic energies that are assumed to represent calculations carried out in an infinite basis of oneparticle functions (the basis-set limit). In practice, finite basis sets are used; as we shall see, the truncation of the one-electron basis is a serious problem that may lead to large errors in the calculations.
As seen from Table 1.5, the convergence with respect to X is slow for the correlation contributions to the AE. Even with the largest basis, we have an error of -3.6 kJ/mol, originating almost exclusively from the basis-set truncation of the doubles contribution to the CCSD energy. The slow convergence arises from the orbital approximation (i.e., the expansion of the wavefunction in determinants), leading to a poor description of the short-range correlated motion of the electrons. Noting that as many as 460 AOs are needed for a small diatomic molecule to achieve chemical accuracy, it is clear that this brute-force approach does not represent a widely applicable tool for the calculation of thermochemical data.
There are two possible solutions to this problem. We may either modify our ansatz for the wavefunction, including terms that depend explicitly on the interelectronic coordinates [26-30], or we may take advantage of the smooth convergence of the correlation-consistent basis sets to extrapolate to the basis-set limit [6, 31-39]. In our work, we have considered both approaches; as we shall see, they are fully consistent with each other and with the available experimental data. With these techniques, the accurate calculation of AEs is achieved at a much lower cost than with the brute-force approach described in the present section.

12 |
Chapter 1 |
5.SHORT-RANGE CORRELATION AND THE COULOMB HOLE
In the preceding section, we observed the slow basis-set convergence of the doubles contributions to the AE of CO. In the present section, we shall make an attempt at understanding the reasons for the slow convergence and to see if this insight can help us design better computational schemes.
5.1.Terms Linear in 
The slow convergence of the doubles contributions to the AE is a general problem related to the accurate description of electron pairs in any electronic system. This problem has been studied carefully for the simplest two-electron system, namely the ground-state He atom. In nonrelativistic theory, its Hamiltonian reads
where (in atomic units) the first two and the last three terms represent the kinetic energies of the two electrons and the Coulomb interactions between the three particles, respectively. As two particles coalesce, the potential part of the Hamiltonian becomes singular in the left-hand side of the Schrödinger equation
For the right-hand side to remain finite, there must be a compensating term arising from the kinetic energy part on the left-hand side. In particular, for the singlet ground state, Slater found that the wavefunction must satisfy the following cusp conditions for coalescing particles [40, 41]:
(spherical averaging implied). These conditions are satisfied if, for example, the wavefunction behaves in the following manner for small interparticle distances:

Highly Accurate Ab Initio Computation of Thermochemical Data |
13 |
Whereas the one-electron exponential form Eq. (5.5) is easily implemented for orbital-based wavefunctions, the explicit inclusion in the wavefunction of the interelectronic distance Eq. (5.6) goes beyond the orbital approximation (the determinant expansion) of standard quantum chemistry since  does not factorize into one-electron functions. Still, the inclusion of a term in the wavefunction containing
does not factorize into one-electron functions. Still, the inclusion of a term in the wavefunction containing  linearly has a dramatic impact on the ability of the wavefunction to model the electronic structure as two electrons approach each other closely.
linearly has a dramatic impact on the ability of the wavefunction to model the electronic structure as two electrons approach each other closely.
To see the importance of the  term, consider the standard FCI expansion of the He ground-state wavefunction. The FCI wavefunction is written as a linear expansion of determinants,
term, consider the standard FCI expansion of the He ground-state wavefunction. The FCI wavefunction is written as a linear expansion of determinants,
each of which contains a product of two Slater-type orbitals (STOs),
where |
is a spherical-harmonic function. The same (optimized) |
|
exponent |
is used for all STOs, which differ only in the quantum num- |
|
bers |
By including in the FCI wavefunction all STOs |
|
up to a given principal quantum number |
a sequence of FCI |
|
wavefunctions is established, which approaches the exact nonrelativistic wavefunction as X tends to infinity. In the following, we shall refer to this hierarchy of FCI wavefunctions as the principal expansion [12].
For |
the principal expansion contains only one determinant (the |
|
Hartree-Fock determinant); for |
the FCI wavefunction is a multi- |
|
determinant expansion. |
|
|
|
To illustrate the convergence of the FCI principal expansion with |
|
respect to short-range electron correlation, we have in Fig. 1.1 plotted the ground-state He wavefunction with both electrons fixed at a distance
of |
from the nucleus, as a function of the angle |
between the |
||
position vectors |
and |
of the two electrons. The thick grey lines |
||
correspond to the exact nonrelativistic wavefunction, whereas the FCI wavefunctions are plotted using black lines. Clearly, the description of the Coulomb cusp and more generally the Coulomb hole is poor in the orbital approximation. In particular, no matter how many terms we include in the FCI wavefunction, we will not be able to describe the nondifferentiability of the wavefunction at the point of coalescence.

14 |
Chapter 1 |
However, this deficiency of the FCI expansion is easily rectified by including in the wavefunction a single extra term that is linear in the interelectronic distance. The resulting wavefunction may be written as [42]
where the coefficients of the CI expansion and of the 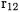 term are optimized simultaneously with the orbital exponent
term are optimized simultaneously with the orbital exponent  . The corresponding wavefunctions are plotted using a dotted line in Fig. 1.1. The improvement in the description of the Coulomb hole is dramatic – already when the
. The corresponding wavefunctions are plotted using a dotted line in Fig. 1.1. The improvement in the description of the Coulomb hole is dramatic – already when the  term is added only to the Hartree-Fock determinant. The improvement in the energy is just as impressive. Whereas the standard FCI principal expansion has errors of 147, 37, 12, and 6.5 kJ/mol for
term is added only to the Hartree-Fock determinant. The improvement in the energy is just as impressive. Whereas the standard FCI principal expansion has errors of 147, 37, 12, and 6.5 kJ/mol for 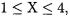 the corresponding errors with the
the corresponding errors with the  term included are only 33, 2.3, 0.39, and 0.14 kJ/mol.
term included are only 33, 2.3, 0.39, and 0.14 kJ/mol.