

Chapter 6
Theoretical Thermochemistry of Radicals
David J. Henry and Leo Radom
Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia
1.INTRODUCTION
In general, radicals are highly reactive species and can therefore often be difficult to study experimentally [1]. Nevertheless, there is a number of experimental procedures that can be used to determine radical thermochemistry, either directly or indirectly (e.g. through thermochemical cycles) [2]. Berkowitz, Ellison and Gutman [3] have reviewed several of these methods and noted their strengths and limitations. Developments in computer technology mean that ab initio molecular orbital theory [4] now provides a viable alternative source of quantitative gasphase thermochemical information [5]. However, the theoretical treatment of open-shell systems such as radicals presents its own difficulties [6]. Therefore, the accurate prediction of radical thermochemistry with theoretical procedures poses an interesting challenge.
In this chapter, we look closely at the performance of several ab initio techniques in the prediction of radical thermochemistry with the aim of demonstrating which procedures are best suited in representative situations. We restrict our attention to several areas in which we have had a recent active interest, namely, the determination of radical heats of formation  bond dissociation energies (BDEs), radical stabilization energies (RSEs), and selected radical reaction barriers and reaction enthalpies. We focus particularly on the results of our recent studies.
bond dissociation energies (BDEs), radical stabilization energies (RSEs), and selected radical reaction barriers and reaction enthalpies. We focus particularly on the results of our recent studies.
161
J.Cioslowski (ed.), Quantum-Mechanical Prediction of Thermochemical Data, 161–197.
©2001 Kluwer Academic Publishers. Printed in the Netherlands.
162 |
Chapter 6 |
2. |
THEORETICAL PROCEDURES |
There is a wide variety of ab initio techniques available for the study of radical thermochemistry, ranging from quite cheap and approximate methods to much more expensive and accurate approaches. The quality of results yielded by these procedures depends on the size of the basis set used and on the degree of electron correlation included. In practice, it is necessary to strike a balance between the required accuracy and the computational cost that can be afforded.
Commonly-used basis sets include those of Pople and coworkers [4] and Dunning and coworkers [7]. The Pople sets range from small basis sets such as 3-21G, to medium-sized basis sets such as 6-31G(d), to large basis sets such as 6-311+G(3df,2p) or G3large. There are several series of Dunning basis sets including cc-pVnZ, aug-cc-pVnZ (containing diffuse functions), and cc-pCVnZ (containing core-correlation functions). These basis sets increase in size as n goes from D to T, to Q, to 5, to 6, etc..
There is also a hierarchy of electron correlation procedures. The Hartree-Fock (HF) approximation neglects correlation of electrons with antiparallel spins. Increasing levels of accuracy of electron correlation treatment are achieved by Møller-Plesset perturbation theory truncated at the second (MP2), third (MP3), or fourth (MP4) order. Further inclusion of electron correlation is achieved by methods such as quadratic configuration interaction with single, double, and (perturbatively calculated) triple excitations [QCISD(T)], and by the analogous coupled cluster theory [CCSD(T)] [8].
Density functional theory (DFT) [9] is becoming increasingly important in determining chemical properties. Typical methods involve the BLYP functional and the hybrid B3LYP procedure. DFT methods are attractive in that they are often highly cost effective and therefore offer the possibility of application to quite large systems, provided that they are suitably reliable.
Apart from the selection of basis set and correlation procedure, an additional consideration arises in open-shell systems because of the presence of one or more unpaired electrons. This leads to treatments that are referred to as spin-restricted (R), spin-unrestricted (U), and spin-projected (P).
Spin-restricted procedures, signified by an R prefix (e.g. RHF, RMP), constrain the  and
and  orbitals to be the same. As such, the resulting wavefunctions are eigenfunctions of the spin-squared operator
orbitals to be the same. As such, the resulting wavefunctions are eigenfunctions of the spin-squared operator  that correspond to pure spin states (doublets, triplets, etc). The disadvantage of this approach is that it restricts the flexibility in the
that correspond to pure spin states (doublets, triplets, etc). The disadvantage of this approach is that it restricts the flexibility in the

Theoretical Thermochemistry of Radicals |
163 |
electronic description and may result in unrealistic spin localization in radicals.
Spin-unrestricted procedures, designated by the prefix U (e.g. UHF, UMP), treat the  and
and  electrons independently. This allows more flexibility in accommodating the unpaired electron(s) and, in the case of the Hartree-Fock wavefunction, often leads to a lower-energy description of the electronic structure. However, treating the
electrons independently. This allows more flexibility in accommodating the unpaired electron(s) and, in the case of the Hartree-Fock wavefunction, often leads to a lower-energy description of the electronic structure. However, treating the  and
and  electrons separately permits the introduction of spin contamination (i.e. mixing of higher spin states) since the wavefunction is no longer an eigenfunction
electrons separately permits the introduction of spin contamination (i.e. mixing of higher spin states) since the wavefunction is no longer an eigenfunction
of |
The degree of spin contamination is reflected in the deviation of |
the |
expectation value from that of a pure spin state (i.e. 0.75 for a |
doublet, 2.0 for a triplet, etc).
A further alternative is to remove the higher-spin states from the unrestricted wavefunction by means of a spin-projection operator. Spinprojected energies are designated by a P prefix (e.g. PHF, PMP).
It is not clear beforehand which of these alternatives is to be preferred. At the HF and MP levels of theory, the differences between them can be substantial. However, at the QCISD(T) and CCSD(T) levels, it has been found that the differences between the restricted and unrestricted energies are generally small [10, 11].
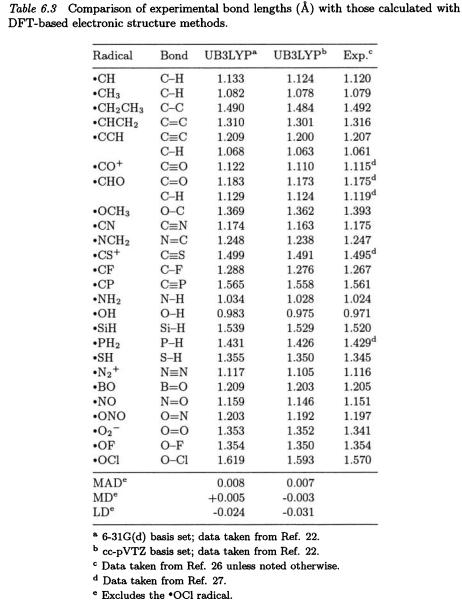
It has been argued [12] that DFT calculations on open-shell systems should always be performed with spin-unrestricted methods. However, it is still of practical interest to compare the performance of procedures such as UB3LYP and RB3LYP in thermochemical predictions [13].
The ideal calculation would use an infinite basis set and encompass complete incorporation of electron correlation (full configuration interaction). Since this is not feasible in practice, a number of compound methods have been introduced which attempt to approach this limit through additivity and/or extrapolation procedures. Such methods (e.g. G3 [14], CBS-Q [15] and  [16]) make it possible to approximate results with a more complete incorporation of electron correlation and a larger basis set than might be accessible from direct calculations. Table 6.1 presents the principal features of a selection of these methods.
[16]) make it possible to approximate results with a more complete incorporation of electron correlation and a larger basis set than might be accessible from direct calculations. Table 6.1 presents the principal features of a selection of these methods.
The Gaussian-n (Gn) methods (e.g. G2 [17], G2(MP2, SVP) [18], G3 [14], and G3(MP2) [19]) attempt to approximate the results of a large basis set UQCISD(T) calculation. A defined series of calculations is performed at the UMP2, UMP4, and UQCISD(T) levels of theory with specific basis sets. Additivity approximations are then used to obtain a molecular energy which, when combined with a scaled zeropoint vibrational energy (ZPVE) and molecule-independent empirical higher-level correction (HLC), gives the Gn total energy at 0 K for the molecule.

164 |
Chapter 6 |
The main feature of the CBS (complete basis set) methods (e.g. CBS-Q [15] and CBS-QB3 [20]) is extrapolation to the complete basis set limit at the UMP2 level. Additional calculations [UMP4 and UQCISD(T) or UCCSD(T)] are performed to estimate higher-order effects. A scaled ZPVE, together with a size-consistent empirical correction and a spin-contamination correction, are added to yield the total CBS energy of the molecule.
Several variations of the Gn and CBS methods have been designed specifically for radicals, and, are therefore labeled with the suffix RAD. The Gaussian-n variants include G2-RAD(QCISD) [21], G2(MP2, SVP)- RAD [21], G3-RAD [22], and G3(MP2)-RAD [23]. These procedures are characterized by the use of alternative geometries and scaled zeropoint energies, replacement of unrestricted open-shell calculations with restricted open-shell methods, and calculation at the highest correlation level with the URCCSD(T) method instead of UQCISD(T) [24]. CBSRAD [25] is a variation of the CBS-Q procedure and makes use of a UB3LYP/6-31G(d) geometry and scaled ZPVE while also replacing the UQCISD(T) calculation with UCCSD(T). The principal features of these variants are also included in Table 6.1.

Theoretical Thermochemistry of Radicals |
165 |
A third class of compound methods are the extrapolation-based procedures due to Martin [5], which attempt to approximate infinite- basis-set URCCSD(T) calculations. In the  method [16] calculations are performed at the URCCSD and URCCSD(T) levels of theory with basis sets of systematically increasing size. Separate extrapolations are then performed to determine the SCF, URCCSD valence-correlation, and triple-excitation components of the total atomization energy at
method [16] calculations are performed at the URCCSD and URCCSD(T) levels of theory with basis sets of systematically increasing size. Separate extrapolations are then performed to determine the SCF, URCCSD valence-correlation, and triple-excitation components of the total atomization energy at
166 |
Chapter 6 |
the basis-set limit. Also included are contributions from core correlation, scaled ZPVE, scalar relativistic effects, and spin-orbit coupling (for atoms only).