


BDEs of Transition Metal Compounds and Main Group Complexes |
203 |
2.HOMOLEPTIC CARBONYL COMPLEXES
The number of TM carbonyl complexes for which experimental BDEs are known is relatively large. Theoretical studies of neutral 18valence electron carbonyl complexes of groups 6, 8 and 10 have been reported [37-42]. Table 7.1 lists the calculated and experimental values of  and
and 
In most cases, the computed values of  are in a very good agreement with experiment. Although the calculated BDEs of
are in a very good agreement with experiment. Although the calculated BDEs of  and
and  are higher than their experimental counterparts, a closer study of the latter suggests that the reported values may be too low. Inspec-
are higher than their experimental counterparts, a closer study of the latter suggests that the reported values may be too low. Inspec-
tion of Table 7.1 leads to the conclusion that the performance of the BP86/II and B3LYP/II levels of theory is very good as the DFT BDEs are close to their CCSD(T) counterparts. The MP2/II data are always too high but the trends in the calculated BDEs are correct. Note that the MP2/II level of theory yields particularly high BDEs for 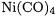
and . These erroneously high values are representative of the problems that are often encountered when first TM row compounds are treated at the MP2 level of theory.

204 |
Chapter 7 |
Table 7.2 lists the calculated and experimental BDEs of two series of charged carbonyl complexes [39, 40, 47]. The first series comprises positively and negatively charged hexacarbonyls  that are isoelectronic with
that are isoelectronic with  There are no experimental bond energies available for
There are no experimental bond energies available for  The B3LYP/II and BP86/II results are very similar to their CCSD(T)/II counterparts while the MP2/II level of theory always yields values of BDEs that are too high. The second set of data consists of theoretical and experimental results for the positively charged group11 carbonyls
The B3LYP/II and BP86/II results are very similar to their CCSD(T)/II counterparts while the MP2/II level of theory always yields values of BDEs that are too high. The second set of data consists of theoretical and experimental results for the positively charged group11 carbonyls  (
(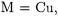 Ag, Au;
Ag, Au;  ). The valence basis sets used for the latter metals were much larger than those employed for the hexacarbonyls. Another difference is that the group-11 cations have a completely filled
). The valence basis sets used for the latter metals were much larger than those employed for the hexacarbonyls. Another difference is that the group-11 cations have a completely filled  shell. The bonding in these compounds has only a negligible
shell. The bonding in these compounds has only a negligible  while backdonation is important in the hexacarbonyls. This different bonding situation leads to an altered performance of theoretical methods. As expected, the CCSD(T) values are in good agreement with experiment. The MP2 values are also quite
while backdonation is important in the hexacarbonyls. This different bonding situation leads to an altered performance of theoretical methods. As expected, the CCSD(T) values are in good agreement with experiment. The MP2 values are also quite

BDEs of Transition Metal Compounds and Main Group Complexes |
205 |
accurate. The BP86 and B3LYP functionals yield bond energies for the monocarbonyls 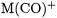 that are much higher than their MP2 and CCSD(T) counterparts. Even more troublesome is the fact that the DFT methods sometimes predict a wrong trend for the BDEs of monoand dicarbonyls. The BP86 and B3LYP functionals predict the BDE of
that are much higher than their MP2 and CCSD(T) counterparts. Even more troublesome is the fact that the DFT methods sometimes predict a wrong trend for the BDEs of monoand dicarbonyls. The BP86 and B3LYP functionals predict the BDE of 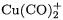 to be lower than that of
to be lower than that of  while MP2 and CCSD(T) levels of theory yield the opposite result, in agreement with experiment.
while MP2 and CCSD(T) levels of theory yield the opposite result, in agreement with experiment.
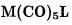
206 |
Chapter 7 |
BP86 also fails to predict the relative BDEs of  and
and  The B3LYP approach produces a higher bond energy of
The B3LYP approach produces a higher bond energy of  as compared with
as compared with  but this difference is much smaller than that predicted within the CCSD(T) approximation.
but this difference is much smaller than that predicted within the CCSD(T) approximation.
3.GROUP-6 CARBONYL COMPLEXES (M = Cr, Mo, W)
Singly substituted species 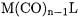 have been investigated [4951, 54, 55]. Table 7.3 lists the calculated BDEs of the group-6 complexes of the type
have been investigated [4951, 54, 55]. Table 7.3 lists the calculated BDEs of the group-6 complexes of the type  with various ligands L, whereas Table 7.4 contains the BDEs for the W–L and the W–CO bonds in
with various ligands L, whereas Table 7.4 contains the BDEs for the W–L and the W–CO bonds in  complexes. The latter values are given for the least bonded carbonyl ligand. Note that BDEs for other
complexes. The latter values are given for the least bonded carbonyl ligand. Note that BDEs for other  complexes with certain particular classes of ligands are discussed in other sections of this chapter.
complexes with certain particular classes of ligands are discussed in other sections of this chapter.
Table 7.3 lists only few experimental data that can be used to estimate the accuracy of the theoretical results. The CCSD(T)/II values agree quite well with experiment (note, however, rather large error bars for the measured BDEs of the thiocarbonyl complexes). The MP2/II values are always larger than their CCSD(T)/II counterparts. The latter values show that the tungsten complexes always have the strongest M–L bond while, in most cases, the molybdenum species have the lowest BDEs.
Table 7.4 lists BDEs calculated with both ab initio and DFT methods. The B3LYP/II values for the  and
and  bond energies are in very good agreement with their CCSD(T)/II counterparts. However, the results yielded by the two methods differ in the relative bond strengths of acetylene and ethylene in the
bond energies are in very good agreement with their CCSD(T)/II counterparts. However, the results yielded by the two methods differ in the relative bond strengths of acetylene and ethylene in the  complexes. The B3LYP/II level of theory predicts ethylene to be less bonded than acetylene while CCSD(T)/II and MP2/II levels yield the opposite result. The unstable
complexes. The B3LYP/II level of theory predicts ethylene to be less bonded than acetylene while CCSD(T)/II and MP2/II levels yield the opposite result. The unstable  and
and  species have been detected experimentally by IR spectroscopy [56]. The decrease in the C–O stretching frequency of the trans CO ligand was found to be significantly larger for the former complex. This observation indicates that the tungsten–acetylene interactions are stronger than the tungsten– ethylene ones, which is in agreement with the larger BDE of
species have been detected experimentally by IR spectroscopy [56]. The decrease in the C–O stretching frequency of the trans CO ligand was found to be significantly larger for the former complex. This observation indicates that the tungsten–acetylene interactions are stronger than the tungsten– ethylene ones, which is in agreement with the larger BDE of 
predicted at the ab initio levels of theory. The only experimental BDE given in Table 7.4 is for the weakly bonded complex  . It agrees well with the CCSD(T)/II result.
. It agrees well with the CCSD(T)/II result.