

Konspekt_lekcii
.pdfДобуток m23 16 називається характеристикою капіляра. Він залежить від діаметра і довжини капіляра, висоти підйому напірної посудини і температури. Ця величина легко визначається експериментально і при постійних параметрах установки є сталою.
Концентрацію речовини можна визначити різними способами.
1. Розрахунковий спосіб полягає у фіксуванні полярограми, визначенні граничного дифузійного струму і розрахунку концентрації за рівнянням Ільковича (4.20), розв'язавши його відносно С:
С |
I |
гр |
|
|
(4.23) |
|
|
|
|
|
|||
605nD 12 m |
23 |
16 |
||||
|
|
|||||
Для використання цього простого способу необхідно крім визначення характеристики капіляра знати коефіцієнт дифузії, точне значення якого не завжди відоме, тому цей метод в аналітичній практиці використовується порівняно рідко.
2. Методи калібрування. У методах калібрування не обов’язково знати величину граничного дифузійного струму (Ігр) в мка. Можна користуватися пропорційною їй величиною висоти полярографічної хвилі (h) в мм. Висоту визначають з графіка, побудованого на міліметровому папері або з діаграмної стрічки самописця. Для використання методу абсолютного калібрування або калібрувальних коефіцієнтів необхідно, щоби розчини з досліджуваною речовиною і стандартні розчини мали однаковий коефіцієнт дифузії (D). Ця умова виконується, якщо до однакових об'ємів фонового електроліта додають однакові об'єми (не більше 10% від об'єму фонового електроліта) досліджуваного і стандартних розчинів.
У цьому випадку всі постійні величини можна об’єднати в одну константу і рівняння (4.22) буде мати вигляд:
h = kC |
(4.24) |
Для аналізу розчинів, концентрація визначуваної речовини в яких лежить на границі чутливості, використовують метод добавок. В електролітичну комірку вміщають певний об’єм суміші досліджуваного розчину з фоновим електролітом (Vx) і вимірюють граничний дифузійний струм (hx). Потім в цей же електролізер додають точно виміряний об’єм концентрованого стандартного розчину (Vст, Сст) і знов вимірюють граничний дифузійний струм (hx+ст). Концентрацію досліджуваного розчину розраховують за формулою:
|
Cx |
CстVстhx |
, |
(4.25) |
||
|
Vx (hx ст hx ) |
|||||
|
|
|
|
|||
або з урахуванням зміни об'єму: |
|
|
|
|
|
|
Cx |
|
CстVстhx |
|
|
(4.26) |
|
((Vx Vст )hx ст |
hxVx ) |
|||||
|
|
|||||
Методом класичної полярографії можна визначати концентрації до 10-5 моль/л, а в деяких |
||||||
випадках до 10-7 моль/л з точністю 2-3%. Широко використовується для аналізу неорганічних і органічних речовин.
Недоліком метода є неможливість аналізу неелектроактивних речовин і необхідність роботи з шкідливою речовиною - ртуттю.
4.2.6. Причини спотворення форми полярограм.
При одержанні та інтерпритації полярограм необхідно враховувати деякі явища, які спотворюють форму полярографічної кривої.
73
Конденсаторний струм зумовлений виникненням подвійного електричного шару, утвореного іонами фону навколо електрода. На полярограмі замість горизонтальних ділянок з’являються ділянки похилі, які зменшують чутливість метода. Сила конденсаторного струму залежить від швидкості підйому потенціалу електрода. Для зменшення конденсаторного струму необхідно зменшити швидкість розгортки потенціалу електрода.
Хвилі кисню з’являються на полярограмі внаслідок відновлення кисню, розчиненого в досліджуваному розчині. Кисень дає дві хвилі. Перша хвиля зумовлена відновленням його до пероксиду водню при потенціалі –0,15...–0,2 В:
O2 2H 2e H2O2 .
Друга хвиля виникає при –0,7...–1,3 В за рахунок відновлення пероксиду водню до води:
H2O2 2H 2e 2H2O .
Для видалення розчиненого кисню через розчин пропускають газ (Н2, N2, Ar). Через кислі розчини можна пропускати СО2, а до лужних розчинів додають 0,1 г/100 мл Na2SO3.
Полярографічні максимуми. Максимуми І роду утворюються на підйомі полярографічної хвилі і мають форму гострих піків. Згідно теорії академіка Фрумкіна, причиною виникнення максимумів І роду є неоднаковість густини струму у різних частинах краплі ртуті. У нижній частині густина струму більша, у верхній менша завдяки екрануванню верхньої частини поверхнею капіляра. Густина струму впливає на поверхневий натяг ртуті. Різниця поверхневого натягу викликає рух поверхні ртуті, що призводить до перемішування розчину у дифузійному шарі і збільшення сили струму. Цей ефект найбільше проявляється при потенціалі –0,56 В. Максимуми І роду з’являються на фоні розбавлених розчинів.
Максимуми ІІ роду виникають при швидкому витіканні ртуті з капіляра у концентрованих розчинах і мають заокруглену форму при всіх потенціалах. Струмінь ртуті доходить до дна краплі, розходиться по боковій поверхні вгору, викликає рух поверхні ртуті і перемішування розчину, що призводить до виникнення максимуму на полярограмі.
Від максимумів І і ІІ роду позбавляються, додаючи до розчину поверхневоактивні речовини, які зменшують і вирівнюють поверхневий натяг, припиняють рух поверхні ртуті і усувають причину зростання струму. Як поверхневоактивні речовини використовують желатин, агар-агар та ін.
Ефект зменшення максимумів І і ІІ роду використовують для визначення концентрації поверхневоактивних речовин з чутливістю до 10-9 моль/л.
Максимуми III і IV роду виникають при використанні твердих електродів.
4.2.7. Амперометричне титрування.
Залежність сили дифузійного струму при відновленні або окисненні визначуваної речовини на електроді від її концентрації в розчині може бути використана для визначення кінця титрування в титриметричному аналізі. Такий метод носить назву амперометричне титрування і належить до непрямих полярографічних методів аналізу.
У 1927 році Я. Гейровський запропонував після кожного додавання титранту знімати полярограму і будувати криву титрування. В 1936 році Майєр показав, що можна фіксувати струм при постійному потенціалі, який відповідає граничному дифузійному струму і знімати залежність його величини від об’єму стандартного розчину.
При проведенні амперометричного титрування на електроди подають постійну напругу, при якій досягається ділянка граничного дифузійнгого струму хоча б для однієї з реаґуючих речовин або продукту реакції.
74
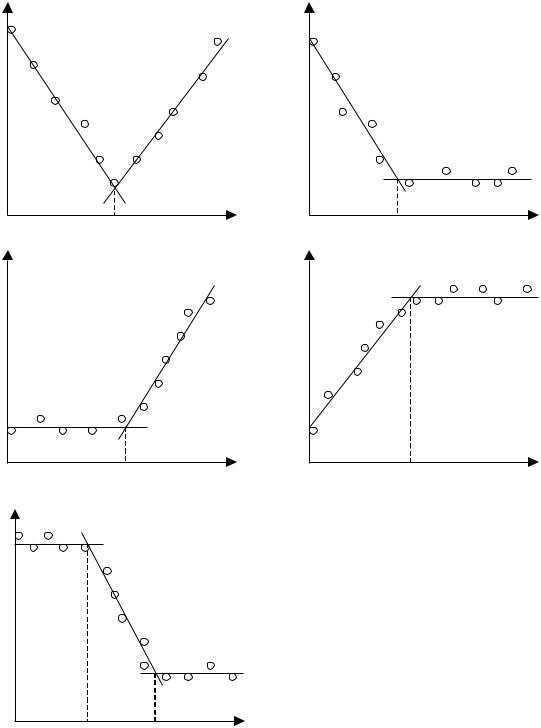
Очевидно, для амперометричного титрування можуть використовуватися реакції, в результаті яких змінюється концентрація електродноактивних, тобто, здатних відновлюватися або окиснюватися на індикаторному електроді, речовин. Це реакції осадження, комплексоутворення або окиснення-відновлення.
Аналітичним сигналом в амперометричному титруванні є об’єм титранту в точці еквівалентності, яку знаходять за різким зломом на кривій залежності сили струму від об’єму прилитого титранту. Вид цієї графічної залежності зумовлений здатністю до електродної реакції як визначуваної речовини, так і титранта або продуктів реакції (рис. 4.8).
I |
|
а |
|
VT.E. |
VT |
I |
|
в |
|
VT.E. |
VT |
I |
|
|
д |
VT.E. |
VT |
I |
|
|
б |
VT.E. |
VT |
I |
|
|
г |
VT.E. |
VT |
Рис. 4.8. Форми кривих амперметричного титрування
75

Якщо при поданій на електролітичну комірку напрузі електродноактивними є визначувана речовина і титрант, то гранична сила струму до точки еквівалентності буде зменшуватися, а за точкою — зростатиме (рис. 4.8а) Така форма залежності має місце, наприклад, при титруванні йонів плюмбуму біхроматом калію:
2 Pb2+ + Cr2O72– + H2O 2 PbCrO4 + 2 H+
У тому випадку, коли електродноактивною при заданій напрузі є тільки визначувана речовина, а електроднонеактивною — титрант, сила струму буде падати до точки еквівалентності, а за точкою — залишатиметься малою і постійною (рис. 4.8б), як, наприклад, при титруванні йонів нікелю диметилгліоксимом (ДМГО):
|
|
|
|
|
H |
|
|
|
|
H3C |
|
|
O |
|
|
H |
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
C |
|
|
|
N |
|
O |
|
N |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
||||||||||||
Ni2+ |
HON |
|
|
|
C |
|
CH3 |
|
|
|
|
|
|
|
|
|
|
|
|
|
CH3 + 2H+ |
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
+ 2 |
|
|
|
|
|
|
|
|
H3C |
|
C |
|
|
|
|
Ni |
|
C |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||
|
HON |
|
CH |
|
CH3 |
|
|
|
|
|
|
||||||||||||
|
|
|
|
|
N |
|
|
O |
|
N |
|
C |
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
H |
|
O |
|
CH3 |
|||
У протилежному випадку, тобто коли електродноактивним буде титрант, а електроднонеактивною — визначувана речовина, сила струму до точки еквівалентності буде залишатися малою та постійною і зростатиме за точкою еквівалентності (рис. 4.8в), як це має місце при титруванні йонів барію біхроматомкалію:
2 Ba2+ + Cr2O72– + H2O 2 BaCrO4 + 2 H+
На рис. 4.8г наведено залежність, що одержується при амперометричному титруванні арсенатної кислоти йодидом калію:
HAsO42– + 2 J– + 2 H+ HAsO32– + J2 + H2O
По мірі відтитровування концентрація вільного йоду, що виділяється як продукт реакції, буде зростати до точки еквівалентності, а за точкою — залишатиметься постійною. Аналогічним чином буде змінюватися сила струму, зумовленого відновленням йоду на катоді.
Часто застосовують так зване індикаторне амперометричне титрування, яке полягає в тому, що точку еквівалентності при титруванні електроднонеактивного йона електроднонеактивним титрантом визначають за зменшенням сили струму, зумовленого відновленням на електроді індикаторного йона, як, наприклад, при титруванні йонів Al3+ розчином фториду в присутності Fe3+. Йон алюмінію утворює з фторидом міцніший комплекс [AlF4]– ( = 7.10), ніж комплекс [FeF4]– ( = 6.10), тому спочатку відтитровуються йони алюмінію і лише коли весь Al3+ відтитрується, починають відтитровуватися йони заліза. Внаслідок цього на кривій залежності сили струму від об’єму титранту спостерігаються два злами (рис. 4.8д), що відповідають точкам еквівалентності для Al3+ та Fe3+.
Точку еквівалентності в кожному випадку знаходять графічно за перетином двох прямих, що відповідають двом ділянкам титрування — до точки еквівалентності та після точки еквівалентності. Слід зауважити, що лінійність між силою струму та об’ємом титранта зберігається лише тоді, коли можна знехтувати розведенням розчину в електролізері, наприклад, у випадку, коли концентрація титранту на порядок більша від концентрації визначуваного компонента, або коли титруючий реаґент ґенерується електрохімічно. У
76
випадку порушення лінійності для побудови графіка титрування слід розрахувати поправки на розведення за формулою:
I |
в ипр |
|
Iв им(Vп Vт ) |
, |
(4.27) |
|
Vп |
||||||
|
|
|
|
|||
|
|
|
|
|
де Івим та Івипр — сила струму виміряна та сила струму виправлена;
Vп та Vт — початковий об’єм розчину та об’єм доданого титранту, мл.
Методом амперометричного титрування можна визначати практично всі елементи періодичної системи та велику кількість органічних сполук. Метод простий і не вимагає складної апаратури.
Основною позитивною якістю методу є висока вибірковість: підбором потенціалу досягають умов, при яких в електрохімічній реакції бере участь лише одна речовина з багатокомпонентної суміші — учасник хімічної реакції. Амперометрично можна титрувати каламутні та забарвлені розчини.
Метод дозволяє проводити визначення малих кількостей речовин в досить розведених розчинах, бо амперометрична індикація кінця титрування є найчутливішою. Нижня межа визначуваних концентрацій досягає 1∙10–6 М.
4.2.8. Інші види полярографії.
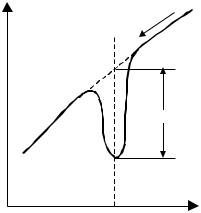
Амальгамна полярографія з накопиченням. У цьому виді полярографії спочатку проводять електроліз розчину визначуваної речовини певний час на стаціонарній ртутній краплі або твердому електроді при потенціалі на 0,2 – 0,3 В більш негативному, ніж потенціал півхвилі визначуваного катіона. Після цього виділений елемент анодно розчиняють при зміні потенціалу з постійною швидкістю від потенціалу електролізу до нуля, фіксуючи при цьому силу струму. Полярограма розчинення має вигляд зубця або оберненого піка (рис. 4.9).
І |
h |
–Е |
Рис. 4.9. Полярограма анодного розчинення визначуваного йона
(стрілкою позначено напрям зміни потенціалу).
Глибина зубця (h) залежить від розмірів краплини ртуті, часу електролізу, швидкості зміни напруги при розчиненні, концентрації визначуваного йона. При постійних умовах експерименту глибина зубця пропорційна концентрації визначуваного йона і дозволяє визначити її методом абсолютного калібрування. Чутливість методу 10-7–10-9 моль/л, точність 5–10 %. Цей метод використовується для визначення домішок в особливо чистих речовинах. Можна визначати декілька катіонів, які відрізняються за потенціалом виділення.
Осцилографічна полярографія. Класична полярографія проводиться при швидкостях зміни потенціалу 0,02 – 0,4 В/хв, амальгамна 1–2 В/хв, а в осцилографічній полярографії швидкість зміни потенціалу на електроді складає десятки вольт за секунду. Це дозволяє
77
збільшити чутливість методу, бо зростає сила струму, але для замірів цього струму, що швидко змінююється, не можуть застосовуватись звичайні магнітоелектричні гальванометри через їх інерційність. Тому для реєстрації використовують електронні прилади – осцилографи, в яких полярограма фіксується на екрані електронно-променевої трубки і має вигляд кривої з піком, максимум якого пропорційний концентрації речовини, а потенціал, який відповідає максимуму піка дорівнює потенціалу півхвилі. Чутливість цього варіанту полярографії 10-5 – 10-6 моль/л.
Зміннострумова полярографія відрізняється від класичної тим, що на електрод разом з лінійною і повільно зростаючою напругою накладається змінна напруга (синусоїдальна або квадратнохвильва) невеликої амплітуди (до 50 мВ). Дифузійний струм при цьому теж містить змінну складову частину, яку можна легко виділити. Це дає можливість зменшити майже до нуля конденсаторний струм, який заважає визначенню малих концентрацій, і завдяки цьому збільшити на порядок (до 10-7 моль/л) чутливість метода. Полярограма при цьому має вигляд, як і в осцилографічній полярографії, де висота піка пропорційна концентрації деполяризатора, а потенціал максимума співпадає з потенціалом півхвилі.
На даний час розроблено інші варіанти, які розширюють можливості полярографічних методів аналізу. Випускаються прилади, які дають можливість реалізувати декілька видів полярографії, наприклад, класичну, зміннострумову і амперометричне титрування.
Питання для самоконтролю.
11.Основи полярографічних методів аналізу.
12.Види поляризації.
13.Принципова схема полярографічної установки.
14.Переваги та недоліки ртутного краплинного електрода.
15.Полярографічна хвиля. Граничний дифузійний струм.
16.Якісний полярографічний аналіз.
17.Способи проведення кількісного полярографічного аналізу.
18.Причини спотворення форми полярограм, полярографічні максимуми і їх подолання.
19.Амперометричне титрування, його можливості.
20.Способи збільшення чутливості полярографічних методів аналізу.
4.3. Кондуктометричні методи аналізу
4.3.1. Електропровідність розчинів електролітів.
Кондуктометричні методи аналізу грунтуються на залежності електричної провідності розчинів електролітів від їх складу.
Електричною провідністю називають здатність речовин проводити електричний струм під дією зовнішнього джерела електричного поля. Носіями зарядів в розчинах електролітів є іони різного заряду: катіони і аніони, які рухаються відповідно до електродів з протилежним зарядом.
Для вимірювання електричної провідності в розчин занурюють два індиферентних електрода, до яких прикладають напругу від джерела електричного струму. Для запобігання зміни складу розчину завдяки електролізу і зменшенню поляризації електродів при вимірюванні електропровідності розчинів електролітів використовують змінний струм. Електрична провідність вимірюється як сила струму, яка проходить через розчин в розрахунку на одиницю прикладеної до електродів напруги, і розраховується за формулою:
78
W |
I |
, |
(4.28) |
|
U |
||||
|
|
|
де W – електрична провідність, См; І - сила струму, А;
U - напруга, В.
Одиницею електричної провідності є Сіменс (См). Електрична провідність обернено пропорційна опору:
W |
1 |
, |
(4.29) |
|
R |
||||
|
|
|
розмірність електричної провідності [См] = [Ом-1].
Електрична провідність розчинів електролітів залежить від розмірів і розташування електродів, температури, природи розчинника, властивостей іонів і їх концентрації.
Електрична провідність прямопропорційна площі електродів (S) і обернено пропорційна відстані між електродами (l):
W æ |
S |
(4.30) |
|
l |
|||
|
|
Коефіцієнт пропорційності æ називається питомою електричною провідністю і дорівнює електричній провідності розчину при вимірюванні з допомогою електродів площею 1 см2, які розташовані на відстані 1 см один від одного:
æ |
W l |
(4.31) |
|
S |
|||
|
|
Відношення l/S називають константою електролітичної комірки.
Питома електрична провідність залежить від температури, розчинника, властивості йонів і їх концентрації. Для дисоціації AB ↔ Az+ + Bz-:
æ CF(z u z u ), |
(4.30) |
де - ступінь дисоцації електроліта. Для сильних електролітів ~ 1, С - молярна концентрація електроліта,
F - число Фарадея,
z+, z- - заряд катіона та аніона,
u+, u- - швидкість руху катіона та аніона при напруженості електричного поля 1 В/см.
Питома електрична провідність, віднесена до числа моль-еквівалентів іонів в 1 см3 розчину, називається еквівалентною електричною провідністю або рухливістю ( ):
|
æ 1000 |
æV , |
(4.33) |
|
С |
||||
|
|
|
де С - концентрація йонів, моль-еквівалентів/літр,
V – розбавлення – це об’єм розчину в см3, який при відстані між електродами 1 см містить 1 моль еквівалентів іонів.
У кондуктометричних методах аналізу вимірюваним параметром, який залежить тільки від складу розчину електроліту, є питома електрична провідність (æ) або електрична провідність (W), виміряна при незмінній константі електрометричної комірки (l/S). Константу електрометричої комірки зазвичай розраховують за формулою 4.31, вимірявши електричну провідність стандартного розчину електроліту з відомою питомою електричною провідністю. Електричну провідність вимірюють кондуктометрами або розраховують за формулою 4.29, вимірявши опір між електродами (R) реохордним містком. В обох випадках до електродів
79

прикладається змінна напруга. Під час вимірювань обидва електроди повинні повністю бути занурені в розчин.
Питома та еквівалентна електрична провідності залежать від властивості іонів та їх концентрації, температури і розчинника.
При низьких концентраціях електролітів зростання концентрації призводить до збільшення кількості носіїв заряду, що пропорційно збільшує електропровідність (рис. 4.10,а).
æ |
λ |
|
а |
|
1 |
|
2 |
|
3 |
|
C |
б |
1 |
2 |
3 |
C |
Рис. 4.10. Залежність питомої електричної провідності (а) і еквівалентної електричної провідності (б) від концентрації електролітів
(1 – HCl, 2 – KOH, 3 – CH3COOH).
При збільшенні концентрації зростання електропровідності відстає від лінійної залежності, що для слабких електролітів пов'язане зі зменшенням ступеню дисоціації ( ), а для сильних електролітів - із збільшенням іонної сили розчину. При великих концентраціях посилюється міжіонна взаємодія і швидкість руху іонів продовжує зменшуватися внаслідок
катафоретичного і релаксаційного ефектів. Катафоретичний ефект полягає в тому, що атмосфера протийонів, яка оточує кожний іон, рухається в протилежному напрямку і гальмує рух іону. Релаксаційний ефект пояснюється тим, що при русі іону його іонна атмосфера руйнується і створюється на новому місці. Ці ефекти призводять до того, що не тільки уповільнюється зростання електричної провідності, але інколи електрична провідність зменшується зі зростанням концентрації.
Збільшення концентрації електролітів призводить до зменшення еквівалентної електроровідності розчину (рис. 4.10,б). Для сильного 1-1 валентного електроліту її можна розрахувати за рівнянням Онзагера:
|
|
|
|
0 А , |
(4.34) |
||
де A - константа, яка залежить від температури, в’язкості розчину і діелектричної проникності розчинника,
- йонна сила розчину.
0 - гранична еквівалентна електрична провідність (гранична рухливість) - еквівалентна електрична провідність при безмежному розведенні.
Гранична рухливість залежить від температури і природи розчинника. Вона визначається екстраполяцією залежності еквівалентної електропровідності від концентрації електроліта до перетину з віссю ординат.
80

Кольрауш встановив, що гранична еквівалентна електропровідність розчину електроліту є адитивною величиною і складається з граничних рухливостей катіонів і аніонів:
0 = + + – (4.35)
Гранична еквівалентна електрична провідність є фізико-хімічною константою іонів у певному розчиннику при певній температурі і наводиться у довідниках.
Граничні рухливості різних іонів у водних розчинах для більшості іонів знаходяться в межах 40 – 70 См·см2. Граничні рухливості H+ та OH– дорівнюють 362 і 205 См·см2 відповідно, що сильно відрізняється від рухливості інших іонів. Ця аномалія пояснюється естафетним механізмом передавання заряду цими іонами з утворенням водневих зв’язків:
H |
|
|
H |
|
+ |
|
H |
|
|
|
|
|
|
|
|
||
H+ + O |
|
H = |
H . . O . . H |
|
= H |
|
O + H+ |
|
|
|
|
||||||
|
||||||||
|
|
|
|
|
|
|
|
|
або
H – O– + H – O – H = │H – O . . H . . O – H│– = H – O – H + –O – H.
З адитивності рухливостей і формули (4.33) можна вивести формулу для питомої
електричної провідності розчину, який містить декілька іонів: |
|
||||
æ |
|
1 |
Сі і |
(4.36) |
|
1000 |
|||||
|
|
|
|||
Електрична провідність і рухливість збільшуються зі збільшенням температури, бо |
|||||
зменшується в’язкість розчинника і збільшується швидкість теплового руху іонів: |
|
||||
t 25 1 a(t 25) |
(4.37) |
||||
де α - температурний коефіцієрт електричної провідності, який залежить від природи іонів і розчинника,
t - температура розчину, оС.
Для водних рочинів цей коефіцієнт дорівнює 0,02 – 0,025. Для точних вимірювань, щоби виключити вплив температури, електрометричну комірку термостатують.
Електрична провідність і рухливість залежать від природи розчинника. А.М. Шкодіним встановлена залежність граничної рухливості від діелектричної проникності і в’язкості розчинника:
|
|
|
А |
|
B |
|
||
|
|
|
|
|
|
|
||
|
0 |
|
e D , |
(4.38) |
||||
|
||||||||
|
|
|
|
|
|
|||
|
|
|
|
|
|
|||
де A i B - константи,
– в’язкість розчинника,
D - діелектрична проникність розчинника.
В неводних розчинах у більшості іонів рухливість менша ніж у водних.
Аналітичний сигнал кондуктометрії (W або æ ) є одномірним і неселективним, його величина залежить від природи і концентрації всіх іонів, які знаходяться в розчині (формула 4.36). Тому якісний аналіз цим методом проводити не можна.
4.3.2 Пряма кондуктометрія.
Кількісний аналіз методом прямої кондуктометрії проводиться в таких випадках:
а). Аналіз бінарних розчинів. Бінарні розчини складаються з двох компонентів - розчинника і одного розчиненого електроліта. Наприклад, вода-H2SO4, вода-NaCl, H2SO4 - SO3 (олеум).
81
б). Аналіз псевдобінарних розчинів. Псевдобінарні розчини містять декілька електролітів, але за умовами їх використання змінюється концентрація одного електроліта, а концентрації інших електролітів постійні. Наприклад, водний розчин NaCl - NaOH.
в). Контроль чистоти розчинника. Якщо чистий розчинник має низьку електричну провідність, наприклад, дистильована вода, а домішки електролітів електричну провідність різко збільшують, значення питомої електричної провідності характеризує чистоту розчинника.
Увипадках а) і б) визначення концентрації електроліта проводять методом абсолютного калібрування, і тільки у випадку б) стандартні розчини необхідно готувати з врахуванням постійних концентрацій інших електролітів.
Увипадку в) заздалегідь визначають, яка питома електрична провідність відповідає необхідній чистоті розчинника. Очистку проводять доти, поки питома електрична провідність очищеного зразка стане меншою визначеного значення. Цей варіант можна використати для контролю очистки стічних вод або для приблизного визначення загальної мінералізації технологічних і природних вод.
Переваги метода прямої кондуктометрії: простота, надійність, достатня точність результатів (1-2%), доступне обладнання. Недоліки: обмеженість об’єктів аналізу, вплив на результати забруднення сторонніми електролітами; при високих концентраціях результати можуть бути не однозначними.
4.3.3. Кондуктометричне титрування.
Ширше застосування дістав метод кондуктометричного титрування, який полягає в побудові залежності електричної провідності розчину від об’єму титранта (кривої титрування) і визначенні точки еквівалентності за зламом на кривій титрування.
Розчин, який аналізують, вміщають в електрометричну комірку з перемішуючим пристроєм (магнітною мішалкою). Розчин титранта однаковими порціями додають в комірку і через деякий час записують електричну провідність розчину. Якщо титрування триває декілька хвилин, комірку можна не термостатувати. Для одержання точніших результатів і при тривалому титруванні електрометричну комірку термостатують. Щоб зменшити зміну електричної провідності розчину за рахунок розбавлення його титрантом, бажано використовувати титрант з більшою концентрацією, який подають з мікробюретки.
Злам на кривій титрування спотерігається, якщо одним з продуктів реакції титрування є малодисоційована сполука або відбувається перетворення іонів в молекулярну форму і навпаки.
Кислотно-основне титрування. При титруванні сильних і слабких кислот та основ
малодисоціюючим продуктом є вода. Наприклад, для сильної кислоти:
H+ + An- + Kt+ + OH- = Kt+ + An- +H2O
До точки еквівалентності в розчині відбувається заміна рухливих іонів водню на малорухливі іони Kt+ і електрична провідність зменшується. Після точки еквівалентності продовжується збільшення концентрації іонів Kt+ і з’являється надлишок іонів гідроксилу, що призводить до збільшення електричної провідності. При титруванні слабких кислот, в залежності від силового показника кислоти, зменшення електричної провідності до точки еквівалентності буде не таким різким, як для сильних кислот, але після точки еквівалентності зростання кривої буде таким самим (рис. 4.11 (а, б, в)).
82