

Konspekt_lekcii
.pdfсортуванні сталей, різних стопів. Для цієї мети розроблені портативні спектроскопи з автономним живленням для проведення аналізу на місці без відбору проб.
Точний кількісний аналіз проводиться при фотографічному та фотоелектричному фіксуванні спектрів. При фотоелектричному фіксуванні спектрів, залежно від стабільності умов збудження, користуються або методом абсолютного калібрування, або методом гомологічних пар.
Фотографічна реєстрація спектрів має деякі особливості. На фотопластинках лінії спектра фіксуються у вигляді тонких, темних смужок, почорніння яких (S) визначається за формулою:
S lg |
I0 |
(2.6.), |
|
I |
|||
|
|
де І0 -інтенсивність світла, яке проходить через незасвітлену ділянку пластинки, І - інтенсивність світла, яке проходить через зображення лінії.
Величина S залежить від експозиції (Н), яка дорівнює добутку інтенсивності лінії (І) на час витримки (t). Ця залежність називається характеристичною кривою фотопластинки
(рис. 2.9).
S |
|
D |
|
C |
|
|
|
|
A |
B |
|
lgH
Рис. 2.9. Характеристична крива фотопластинки.
На кривій можна виділити 3 ділянки. Ділянки AB i CD характеризуються малою залежністю почорніння від lgH, і називаються ділянками недотримки і перетримки, відповідно. Ділянка CD характеризується практично лінійною залежністю почорніння від lgH і називається ділянкою нормального почорніння. При постійному часі витримки ділянку нормального почорніння можна виразити формулою:
S lg I j (t=const)
де - коефіцієнт контрастності; j – коефіцієнт інерційності.
У методі гомологічних пар фіксують почорніння лінії визначуваного елемента-стандарта (Sст). Для них можна записати:
Sx lg I x j Sст lg Iст j
Віднімаючи друге рівняння від першого одержимо:
|
|
|
|
|
|
|
|
|
|
|
I |
x |
|
S |
x |
S |
ст |
(lg I |
x |
lg I |
ст |
); S |
x |
lg |
|
. |
|
|
|
||||||||||||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
Iст |
||
(2.7),
елемента (Sx) і
(2.8)
Відношення інтенсивностей ліній можна записати через рівняння Ломакіна врахувавши, що лінії гомологічні і Cст =const:
I x |
|
axCxb |
a' |
C b ; |
lg |
I x |
lg a' b lg C |
x |
Iст |
' |
x |
x |
|
Iст |
|||
aстCстb |
|
|
|
|
||||
23
Підставляючи останній вираз у формулу (2.8), одержимо:
S |
x |
(lg a' b lg C |
) A B lg C |
. |
(2.9) |
|
x |
x |
|
|
Ця формула вказує на те, що калібрувальний графік в координатах Sx - lgCx є прямою лінією і використовуєься для визначення Cx.
Оскільки почорніння ліній на фотопластинці залежить не тільки від експозиції, але і від якості фотоматеріалу, складу розчинів і умов обробки фотопластинки, аналіз може проводитися трьома методами.
1. Метод трьох еталонів. Спектри трьох етолонів і декількох зразків фотографують на одну фотопластинку, обробляють її, фотометрують гомологічні лінії еталонів і зразків. За даними еталонів за трьома точками будують калібрувальну пряму, за даними Sx і графіком знаходять lgCx і Cx . Метод точний (1-2 % відн.), але трудомісткий бо вимагає фіксування спектрів трьох еталонів і побудови калібрувального графіка для кожної пластинки.
2.Метод твердого (постійного) графіка. З кожної партії фотопластинок вибирають 1- 2 % від загальної кількості і на них фіксують тільки спектри всіх еталонів. Обробляють пластинки в приблизно однакових умовах, фотометрують гомологічні лінії і будують калібрувальний графік з врахуванням всіх зафіксованих спектрів. На інші пластинки цієї ж партії фотографують тільки спектри зразків, обробляють їх в приблизно тих же умовах, як і пластинки з еталонами і, користуючись побудованим графіком, розраховують концентрацію елемента в зразках. Метод вимагає менших витрат часу і матеріалів, але менш точний (5-10 % відн.) через не повну відтворюваність умов обробки фотопластинок.
3.Метод одного еталона. Це компромісний метод, який грунтується на тій властивості
фотографічного процесу, що при Ix/Iст=1 Sx=0 не залежно від умов обробки і властивостей фотопластинки. Тому калібрувальні прямі, які відрізняються умовами обробки, перетинаються в одній точці на осі lgC (рис. 2.10).
S
lgC
Рис. 2.10. Калібрувальні графіки одержані для різних пластинок.
Координати цієї точки (lgCo, Sо=0) можна розрахувати за перетином калібрувальної прямої будь-якої пластинки з віссю lgC. Калібрувальну пряму пластинки, обробленої в інших умовах, можна побудувати за координатами заздалегідь визначеної точки перетину і даними одного еталона, експонованого на цій пластинці. За еталон обирають зразок, концентрація якого найбільше відрізняється від Со.
Метод має близьку до першого методу точність, але вимагає меншого часу і матеріальних витрат.
2.2.5. Полум'яно-фотометричний аналіз.
Спектральні прилади, призначені для використання газового полум'я як джерела збудження, називаються полум'яними фотометрами. Особливістю методу полум'яної фотометрії є порівняно низька температура газового полум'я. Внаслідок цього збуджуються і
24
випромінюють атоми не всіх елементів. Ті ж атоми, що збуджуються (переважно атоми лужних і лужноземельних елементів), випромінюють не всі можливі лінії. Тому спектр випромінювання містить малу кількість ліній і немає необхідності використовувати диспергуючий елемент з великою розділювальною здатністю. Достатньо між полум'ям і рецептором розташувати світлофільтр, який пропускав би випромінювання визначуваного елемента в області, де немає випромінювання інших елементів. Використання світлофільтрів дозволяє відмовитися від оптичної системи і щілини, що спрощує конструкцію приладів і збільшує їх чутливість.
Стабільність полум'я, як джерела збудження дозволяє користуватися простим і точним методом прямого калібрування.
Об'єкт аналізу вводиться у полум'я у вигляді розпорошених крапель розчину. Тому об'єкт аналізу для фотометрії полум'я необхідно попередньо обробити так, щоб визначуваний елемент перейшов у розчин. Полум'яний фотометр може працювати у безперервному режимі і використовуватися для безпосереднього аналізу рідких потоків. Сконструйовані полум'яні фотометри для одночасного визначення концентрацій декількох елементів.
* * *
Характеризуючи в цілому атомно-емісійний аналіз можна констатувати, що метод має такі переваги:
1.Висока чутливість елементного аналізу (10-3 -10-4 % без збагачення).
2.Документальність (при фотографічному фіксуванні).
3.Універсальність (можна визначати всі елементи).
4.Здатність до автоматизації.
5.Можливість проведення локального аналізу.
Разом з тим слід відмітити недоліки методу:
1.Коштовне обладнання універсального призначення (прилади оптики і точної механіки).
2.Необхідність високої кваліфікації персоналу.
3.При несерійних аналізах вимагає великих витрат часу.
2.3. Коротка характеристика атомно-абсорбційного аналізу
Атомно-абсорбційний аналіз (ААА) грунтується на резонансному поглинанні характеристичного випромінювання елемента його незбудженими атомами, які знаходяться у вільному стані.
Цей метод запропонований австралійським вченим Уолшем у 1956 р.
У цьому методі об'єкт аналізу піддають дії високих температур, щоб перевести його складові частини в стан атомної пари, але не достатній для збудження одержаних атомів. Через шар атомізованого зразка пропускають випромінювання від джерела безперервного спектра. При цьому вільні атоми будуть поглинати фотони, енергія яких відповідає енергії резонансних переходів. На виході випромінювання збіднюється хвилями, які поглинаються атомами зразка, тобто спостерігається спектр поглинання.
Аналітичним сигналом ААА є спектр поглинання випромінювання атомами елементів. Довжина хвилі лінії, яка поглинається, є якісною характеристикою аналітичного сигналу. Ступінь послаблення інтенсивності випромінювання цієї хвилі залежить від концентрації елемента і є кількісною характеристикою цього методу
25
Прилади ААА складаються з таких частин: джерела випромінювання, атомізатора, монохроматора, рецептора.
1. Джерело випромінювання повинно мати в своєму спектрі лінії, довжини хвиль яких дорівнюють резонансним переходам атомів, які визначають. Використовуються такі джерела:
а) Газорозрядні лампи високого тиску (водневі, ртутні), які мають безперервний спектр у видимій і ультрафіолетовій областях. За їх допомогою можна визначати різні елементи.
б) Лампи з порожнистим катодом - це газорозрядні лампи заповнені інертним газом і катодом, виготовленим з металу, який визначають. Лампи мають кварцеве вихідне вікно, через яке виходить випромінювання, що містить інтенсивну резонансну лінію визначуваного елемента. Для визначення кожного елемента необхідно мати відповідну лампу з порожнистим катодом.
в) Лазерне випромінювання. Лазер дозволяє одержувати потужне випромінювання певної довжини хвилі. Зручно використовувати лазери, довжину хвилі випромінювання яких можна змінювати.
2.Призначення атомізатора - перевід елементів зразка в стан атомної пари. Використовуються такі принципи атомізації:
а) Полум'яні атомізатори використовують високу температуру полум'я різних горючих газів з окисниками. Досліджуваний зразок переводиться в розчин, переважно водний, який у вигляді дрібних крапель вводиться у газовий потік перед пальником. Газове полум'я характеризується високою стабільністю умов атомізації і малим фоновим випромінюванням.
б) Електротермічні атомізатори, запропоновані Львовим Б.В. в 1961 р., є графітовими трубчастими електропечами, які можуть нагріватися до 3000 оС. Об'єкт аналізу у вигляді розчину або твердої речовини вміщають на внутрішню холодну стінку печі, включають нагрів, який забезпечує випаровування і атомізацію зразка. Після атомізації через відкриті кінці печі пропускають випромінювання від джерела світла. Для відтворюваності умов атомізації режим нагріву регулюють за програмою компьютера.
3.Монохроматори служать для виділення з випромінювання, яке пройшло через атомізатор, резонансної лінії визначуваного елемента. Використовуються призменні або дифракційні монохроматори з вхідною щілиною і відповідною оптичною системою.
При використанні ламп з порожнистим катодом відпадає необхідність у спеціальному монохроматорі, достатньо використати світлофільтр для відокремлення фонового випромінювання атомізатора. У цьому випадку схема прилада суттєво спрощується.
4.Як рецептор використовують фотоелектричні перетворювачі: фотоелементи, фотопомножувачі, які видають електричний сигнал пропорційний інтенсивності світла, що на них падає.
Якісний аналіз можна проводити на приладах з безперервним спектром освітлення і монохроматором, які дають можливість зареєструвати спектр поглинання атомізованого зразка. Наявність поглинання, яке відповідає довжинам хвиль резонансних ліній свідчить про наявність відповідних елементів. При використанні ламп з порожнистим катодом можна говорити про наявність або відсутність елемента, для визначення якого служить ця лампа.
Кількісний аналіз грунтується на залежності ступеня поглинання випромінювання від концентрації визначуваного елемента. Поглинання монохроматичного випромінювання атомами відбувається за законом, аналогічним закону Бугера-Ламберта-Бера, який ми будемо розглядати в молекулярно-абсорбційному аналізі:
I I |
0 |
10 kcl |
(2.10), |
|
|
|
26
де Io - інтенсивність падаючого світла,
I - інтенсивність світла, яке пройшло через шар атомів,
с - концентрація поглинаючих атомів, пропорційна концентрації елемента в зразку, k - коефіцієнт поглинання,
l - товщина поглинаючого шару.
Коефіцієнт поглинання залежить від геометричних розмірів атомізатора і від умов атомізації (температура, швидкість подачі зразка в атомізатор, наявність інших елементів).
Величину A = lgIo/I називають абсорбційністю. З врахуванням формули (2.8) A = kсl. Прилади ААА дозволяють підтримувати стабільними умови атомізації і геометричні
розміри атомізатора, тому кількісний аналіз проводиться методом прямого калібрування, якщо матриця стандартних розчинів відповідає складу досліджуваного розчину. При неможливості відтворити матрицю користуються методом добавок.
У сучасних приладах сигнал рецептора подається безпосередньо на комп'ютер, де і обробляється згідно заданої програми.
Переваги метода: висока чутливість (0,0001 - 0,0005 мг/л), висока точність (0,5 – 1 %) навіть для великих концентрацій визначуваної речовини.
Питання для самоконтролю
1.Характеристики випромінювання електромагнітних коливань з точки зору хвильової і квантової теорій.
2.Співвідношення між різними характеристиками електромагнітних коливань.
3.Характеристики піддіапазонів оптичного діапазону.
4.Що таке спектр випромінювання?
5.Які бувають спектри випромінювання?
6.На чому грунтуються емісійні спектральні методи аналізу?
7.Механізм випромінювання електромагнітних коливань атомами.
8.Чому випромінювання атомів має лінійчастий характер?
9.Що таке енергія збудження лінії?
10.Ймовірність переходу електронів, які супроводжуються випромінюванням. Правила відбору.
11.Закономірності випромінювання атомів.
12.Аналітичний сигнал емісійної спектроскопії. Якісна і кількісна характеристика аналітичного сигналу.
13.Характеристика основних елементів спектральних приладів.
14.Якісний емісійний спектральний аналіз. Останні лінії.
15.Способи визначення довжини хвилі.
16.Від чого залежить інтенсивність спектральної лінії?
17.Процес самопоглинання.
18.Умови застосування методу прямого калібрування.
19.Метод гомологічних пар. Вимоги до гомологічних ліній.
20.Методи напівкількісного спектрального аналізу. Сфера їх застосування.
21.Фотографічний спосіб фіксування інтенсивності ліній. Характеристична крива фотопластинки.
22.Різновиди методу гомологічних пар при фотографічному фіксуванні спектрів. Переваги і недоліки кожного методу.
23.Особливості полум'яно-фотометричного аналізу.
27
24.Загальна характеристика атомно-емісійного аналізу.
25.Основи атомно-абсорбційного аналізу. Аналітичний сигнал.
26.Основні елементи атомно-абсорбційних приладів.
27.Переваги та недоліки лампи з порожнистим катодом як джерела опромінювання.
28.Від чого залежить ступінь поглинання лінії визначуваного елемента.
29.Загальна характеристика і сфери застосування ААА.
2.4.Молекулярно-абсорбційні методи аналізу.
2.4.1. Механізм і характеристики поглинання електромагнітних коливань молекулами. Аналітичний сигнал.
Молекулярно-абсорбційні методи аналізу грунтуються на поглинанні електромагнітних коливань оптичного діапазону молекулами досліджуваної речовини.
Поглинання електромагнітних коливань здійснюється тільки тоді, коли енергія фотона дорівнює різниці енергій двох енергетичних рівнів молекули.
Розглянемо структуру енергетичних рівнів молекули, яка складається з атомів, пов'язаних між собою хімічними зв'язками.
Енергія молекул складається з:
1. Енергій оптичних (валентних) електронів, які можуть знаходитися або на нижчих
(незбуджених) енергетичних рівнях, або на одному із збуджених рівнів:
Еe Еі Е0 |
(2.11) |
2. Енергії коливання атомів. Розрізняють декілька видів коливань:
а) Валентні - зумовлені періодичною зміною відстані між атомами по лінії, яка їх з'єднує. Якщо розглядати двоатомну молекулу як гармонічний осцилятор, можна розрахувати частоту таких коливань:
|
1 |
|
F |
|
, |
(2.12) |
2 |
|
|
||||
|
|
|
|
|
де - частота коливань, F - силова константа,- приведена маса.
Енергія валентних коливань Ек = h (v + 1/2), де v - коливальне квантове число. Енергія коливальних рівнів ніколи не дорівнює 0.
Для триатомних молекул можливі 2 види валентних коливань без зміни валентного кута
а |
б |
в |
Рис. 2.11. Схема валентних коливань:
а – двоатомних молекул; б, в – триатомних молекул.
б) Для багатоатомних молекул можливі коливання із зміною валентних кутів - деформаційні. Деформаційні коливання бувають 4-х видів.
28

+
ножичкові |
маятникові |
крутильні |
віяльні |
Рис.2.12. Схема деформаційних коливань
в) Як правило, зміна валентного кута супроводжується зміною міжатомних відстаней. Такі коливання називаються валентно-деформаційними.
3. Енергія обертання молекули як цілого навколо центра мас.
Еоб |
h2 |
|
( j 1) j, |
(2.13) |
||
8 |
2 I |
|||||
|
|
|
||||
|
|
м |
|
|
||
де Iм - момент інерції молекули, який залежить від маси атомів і міжатомних відстаней, |
|
|||||
j - обертальне квантове число. |
|
|
|
|
|
|
Енергія молекули становить суму всіх видів енергій. |
|
|||||
Eм Ee |
Ek |
Eоб |
(2.14) |
|||
Найбільшу величину енергії мають електронні збуджені рівні. Коливальні рівні мають меншу енергію, обертальні - ще меншу:
Ee > Ek > Eоб
1000 : 100 : 1
Оскільки величина енергетичних рівнів молекул залежить від її будови, аналітичним сигналом в молекулярно-абсорбційному аналізі є сукупність енергій фотонів, які різні молекули здатні поглинати, тобто спектр поглинання електромагнітних коливань.
Кількісно поглинання світла вимірюється відношенням інтенсивності світла, яке пройшло через шар речовини (Іt ), до інтенсивності падаючого світла (Іo ) вираженим у відсотках:
T |
I t |
100% . |
(2.15) |
|
I 0 |
||||
|
|
|
Ця величина називається прозорістю або пропусканням.
Поглинання у видимому і ультрафіолетовому діапазонах зумовлене електронними переходами, а в інфрачервоному і мікрохвильовому діапазонах - коливальними і обертальними переходами в основному (незбудженому) електронному стані.
В абсорбційній спектроскопії для характеристики поглинання використовувують хвильове число N = ' = 1/ = /c [см-1 ]. Оптичний діапазон поділяється на вужчі піддіапазони:
Діапазон |
Довжина хвилі, нм |
Хвильове число, см-1 |
29

Дальній ІЧ |
106 |
– |
5 ּ 104 |
|
10 |
– |
200 |
Середній ІЧ |
5ּ104 |
– |
2,5 ּ |
|
200 |
– |
4000 |
Ближній ІЧ |
2,5 ּ 103 |
– |
103 |
|
4000 |
– |
13000 |
Видимий |
760 |
– |
760 |
13000 |
– |
28000 |
|
Ближній УФ |
360 |
– |
360 |
28 |
ּ 103 |
– |
50 ּ 103 |
Дальній УФ |
200 |
– |
200 |
5 |
ּ 104 |
– |
106 |
|
|
|
10 |
|
|
|
|
Найбільш інформативними з точки зору хімічного аналізу є середній ІЧ, видимий і ближній УФ діапазони.
На відміну від лінійчастих спектрів поглинання атомів, спектр поглинання молекул має смугастий характер, тобто складається з сукупності більш-менш розмитих смуг розділених ділянками практичної відсутності поглинання.
Причиною розмиванни спектрів поглинання молекул є те, що електронні і коливальні рівні розщеплюються на коливальні та обертальні підрівні і поглинання супроводжується появою в спектрі великої кількості ліній з близькими довжинами хвиль, які, внаслідок обмеженості розділювальної здатності спектральних приладів, зливаються в смуги поглинання. Внаслідок різного співвідношення енергій електронних, коливальних і обертальних ліній смуги поглинання в УФ діапазоні більш розмиті, а в ІЧ діапазоні складаються з порівняно гострих піків.
Таким чином, аналітичним сигналом є двомірна залежність поглинання від частоти або хвильового числа. Параметрами аналітичного сигналу є частота або хвильове число в мінімумі пропускання (N1, N2,...) і інтенсивність поглинання електромагнітних коливань певного хвильового числа.
Сукупність частот, при яких відбувається інтенсивне поглинання світла, залежить від будови і не залежить від кількості молекул і використовується для якісного аналізу.
Залежність інтенсивності поглинання світла певної частоти від кількості або концентрації молекул досліджуваної речовини використовується для кількісного аналізу.
2.4.2. Якісний аналіз.
Якісний аналіз за спектрами поглинання грунтується на таких їх властивостях:
1.Немає двох речовин, які б мали абсолютно однаковий спектр поглинання. Тому якісний аналіз (ідентифікацію) речовин проводять шляхом порівняння спектра досліджуваної речовини із спектрами відомих індивідуальних речовин, одержаних в однакових умовах.
2.Число смуг поглинання залежить від числа активних коливань в молекулі. Активними є коливання, які призводять до зміни дипольного момента молекули. Чим більше атомів в молекулі, тим більша кількість активних коливань.
3.Експериментально досліджено, що деякі функціональні групи в складі молекул мають характерні смуги поглинання великої інтенсивності, які мало залежать від загальної будови молекул. Такі смуги поглинання називають характеристичними або груповими.
Наприклад, якщо органічні молекули містять у своєму складі функціональні групи:
|
|
|
|
|
|
|
|
|
|
|
O |
|
H ; |
|
C |
|
H ; C |
|
O |
||
|
|
|
|
|||||||
|
|
|
||||||||
|
|
|
|
|||||||
30
в спектрах поглинання будуть зафіксовані смуги поглинання при таких значеннях хвильових чисел: 3600-3800 ; 3000 та 1720-1780 см-1, відповідно. Аналогічно для неорганічних сполук - наявність груп =Si=O i –O–Si–O–Si–O– призводить до фіксації смуг поглинання з хвильовими числами 785-800 та 480-515 см-1 .
З допомогою характеристичних коливань можна проводити молекулярний, функціональний, а в деяких випадках, і фазовий аналіз.
Зміщення частоти характеристичних коливань дає інформацію про структуру молекули, про внутрішньомолекулярні або міжмолекулярні взаємодії. Таким чином, вивчення спектрів поглинання дає інформацію як про якісний склад, так і про структуру молекул.
Слід відмітити, що через велику кількість органічних і неорганічних речовин і порівняно малий набір функціональних груп, зробити однозначний висновок про якісний склад об'єкту аналізу тільки за даними спектра поглинання важко. Тому молекулярно-абсорбційний метод часто комбінують з іншими фізико-хімічними методами або з попереднім розділенням об'єкта аналізу на чисті компоненти або простіші суміші.
2.4.3. Закон Бугера-Ламберта-Бера.
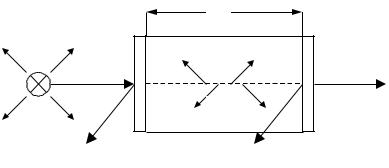
Розглянемо кількісні закономірності поглинання монохроматичного випромінювання шаром речовини (рис.2.13)
|
l |
I0 |
It |
|
Ia |
Ir |
Ir |
Рис. 2.13. Проходження променя світла через шар рідини.
При проходженні світла інтенсивністю Io через шар речовини, вміщеної в кювету з прозорого матеріалу, частина випромінювання (Ir) відбивається або розсіюється на поверхні розділу фаз, частина поглинається, витрачаючись на збудження молекул аналізованої речовини (Ia), решта (It) виходить з кювети:
I0 Ir Ia It
Вибирають такі матеріали і геометрію кювети, щоб Ir < Ia. Для цього матеріал кювети має бути прозорим до падаючого світла, стінки кювети, через які світло заходить і виходить з кювети, повинні бути тонкими і плоскопаралельними. Тоді:
I0 Ia It
Бугер і Ламберт встановили, що однорідні шари однакової товщини (l) поглинають одну й ту ж частку падаючого світлового потоку. Математичним виразом такої залежності є експонента:
I |
t |
I |
0 |
10 kl , |
(2.16) |
|
|
|
|
де l - товщина шару поглинаючої речовини,
k - коефіцієнт поглинання, який для індивідуальних речовин залежить від природи речовини, її концентрації, довжині хвилі падаючого світла і температури.
31

Бер встановив, що коефіцієнт поглинання пропорційний концентрації речовини. |
|
||||
k c |
(2.17) |
||||
Об'єднаний закон Бугера-Ламберта-Бера (Б-Л-Б) має вигляд: |
|
||||
I |
t |
I |
0 |
10 cl |
(2.18) |
|
|
|
|
||
Величину A = lgIo/It називають оптичною густиною або абсорбційністю. З |
|||||
використанням абсорбційності закон Б-Л-Б має вигляд: |
|
||||
A cl |
|
(2.19) |
|||
Коефіцієнт - називають молярним коефіцієнтом поглинання або екстинкцією.
Чисельно він дорівнює абсорбційності зразка товщиною 1см при концентрації 1 моль/л. Екстинкція не залежить від товщини шару і концентрації поглинаючої речовини, а залежить від будови речовини, хвильового числа світла, що проходить через нього, температури і є фізикохімічною константою речовини. Чисельні значення екстинкції, які використовуються в аналізі, в основному лежать в межах 10 –105.
2.4.4. Відхилення від закон Бугера-Ламберта-Бера.
Практика показує, що закон Бера не завжди виконується, тобто спостерігається не лінійна залежність коефіцієнта поглинання від концентрації. Причини відхилення від закону Бера можуть бути наступні:
2. Немонохроматичність випромінювання.
2.Вплив сторонніх речовин (зміщення максимумів поглинання під дією молекул домішок або розчинника).
3.Перебіг у розчині реакцій дисоціації забарвлених речовин:
AB 
 A + B
A + B
4. Гідроліз забарвлених речовиин, ступінь якого залежить від концентрації
[Cu(NH3)4]2+ + 2H2O 
 [Cu(NH3)3H2O]2+ + NH4OH
[Cu(NH3)3H2O]2+ + NH4OH
5. Недостатня стабільність забарвлених комплексів
FeSCN2+ |
|
|
|
|
|
|
|
Fe3+ + SCN– |
|
|
|
||||||
|
|
|
|
|
||||
червон. |
|
|
|
|
|
|
|
Б/барвн. |
Якщо немає надлишку іонів SCN–, рівновага може бути зсунена праворуч. |
||||||||
6. Вплив рН середовища. Під впливом йонів H+ можливий зсув рівноваги в бік утворення |
||||||||
іншої сполуки: |
|
|
|
|
|
|
|
|
2CrO42– + 2H+ |
|
|
|
|
|
|
|
Cr2O72– + H2O |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
||
Для того, щоб провести аналіз, необхідно одержати спектр поглинання об'єкту аналізу, тобто залежність Т або А від N або v´.
2.4.5. Схема приладів для вимірювання спектра поглинання.
Кожний абсорбційний спектральний прилад містить наступні необхідні частини:
1 |
2 |
3 |
|
|
4 |
5 |
||||||
Джерело |
|
|
Моно- |
|
|
Зразок |
|
|
Детектор |
|
|
Реєстратор |
|
|
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|
||||
світла |
|
|
хроматор |
|
|
|
|
|
|
|
||
|
|
|
|
Порівняння |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Рис. 2.14. Принципова схема молекулярно-абсорбційного приладу. |
||||||||||||
32