

Konspekt_lekcii
.pdfW |
W |
|
W |
а |
|
б |
|
Vт.е. |
V(Т) |
Vт.е. |
V(Т) |
|
W |
в |
|
Vт.е. |
V(Т) |
г |
V1т.е.V2т.е. V(Т) |
Рис. 4.11. Криві кондуктометричного титрування сильної кислоти (а), кислоти середньої сили (б), слабкої кислоти (в), суміші сильної і слабкої кислоти (г).
При титруванні суміші слабкої і сильної кислоти можливе фіксування двох точок еквівалентності, якщо різниця силових показників кислот перевищує 4 (рис. 4.11г). Спочатку титрується сильна кислота до першої точки еквівалентності. Витрата титранта між першою і другою точкою еквівалентності відповідає титруванню слабкої кислоти.
Метод осадження. При утворенні малорозчинних сполук до точки еквівалентності теж
відбувається заміна одних іонів іншими при однаковій їх концентрації. Наприклад:
Na+ + Cl- + Ag+ + NO3- = AgCl↓ + Na+ + NO3-
До точки еквівалентності іони хлору замінюються на іони нітрату, які мають дещо меншу рухливість, і електропровідність буде дещо зменшуватись. Після точки еквівалентності, завдяки збільшенню концентрації іонів срібла і нітрату, електропровідність буде суттєво збільшуватись.
Якщо в розчині присутні 2 іони, які утворюють малорозчинні сполуки з титрантом і відношення добутків розчинності цих іонів більше 104, то при суттєвій відмінності рухливостей протиіонів теж можлива фіксація двох точок еквівалентності.
Метод комплексоутворення. Утворення комплексних сполук супроводжується зменшен-
ням кількості іонів в розчині і, відповідно, зменшенням електропровідності, наприклад: 2Cl- + Hg2+ = [HgCl2]
Al3+ + 6F- = [AlF6]3-
Після точки еквівалентності електрична провідність збільшується через збільшення надлишку титранта.
При використанні найбільш широковживаного метода комплексонометрії відбуваються
такі реакції: |
|
до т.е. |
Men+ + H2Y2- = [MeY]n-4 + 2H+, |
після т.е. |
H+ + H2Y2- = H3Y-. |
До т.е. кількість зарядів не змінюється, але з’являються більш рухливі іони водню і електрична провідність збільшується. Після т.е. іони водню зв’язуються з надлишком комплексону і електрична провідність зменшується.
Якщо комплексонометричне титрування проводять у присутності буферних розчинів, точку еквівалентності можна визначати при відмінності рухливостей іонів, які зв’язуються в комплекс і які накопичуються після точки еквівалентності. Якщо іони реагують з комплексоном при різних значеннях pH, вони можуть бути окремо визначені в одному розчині.
Окисно-відновне титрування.Окисно-відновне кондуктометричне титрування практично неможливе, якщо реакція проводиться в сильно кислому або в сильно лужному середовищі (перманганатометрія, біхроматометрія). У цих випадках розчин має велику електричну
83
провідність, яка в процесі титрування мало змінюється. Якщо ж реакція відбувається з достатньою швидкістю в середовищі близькому до нейтрального, зміна електричної провідності в точці еквівалентності стає достатньою для її фіксування.
Наприклад, при йодометричному визначенні арсенітів в присутності соди проходять такі
реакції:
AsO32- + I2 + H2O = AsO43- + 2H+ +2I-
2H+ + 2HCO3- = H2CO3
В результаті першої реакції утворюються рухливі іони I- (0 = 79), а іони H+ зв’язують мало рухливі іони HCO3- (0 = 44) і до точки еквівалентності електропровідність розчину збільшується. Після точки еквівалентності надлишок титранту (спиртовий розчин І2) не змінює електропровідності.
Точність кондуктометричного титрування залежить від характеру зламу кривої титрування в точці еквівалентності. При титруванні сильних кислот та основ до і після точки еквівалентності електрична провідність змінюється практично лінійно, і точка еквівалентності точно визначається за перетином цих прямих. В інших випадках поблизу точки еквівалентності спостерігається більш-менш плавний перехід зумовлений недостатньо великим значенням константи рівноваги реакції титрування. В цьому випадку точку еквівалентності визначають за перетином прямих, проведених через точки досить віддалені від точки еквівалентності.
Кондуктометричне титрування легко піддається автоматизації. Зазвичай використовується подача титранта з постійною витратою і фіксуванням кривої титрування на діаграмній стрічці реєстратора. Точка еквівалентності титрування фіксується за часом, який пройшов від початку титруванні до зламу на кривій титрування. Такий варіант аналізу називається хронокондуктометричним титруванням.
Перевагою кондуктометричного титрування є можливість проведення аналізу каламутних та забарвлених розчинів, використання різних титриметричних методів, визначення органічних і неорганічних сполук, диференційованного титрування сумішей декількох компонентів без розділення. Нижня межа визначуваних концентрацій - 10-4 моль/л при точності не менше 2 %.
4.4. Кулонометричні методи аналізу
4.4.1. Закони електролізу.
Кулонометричні методи аналізу грунтуються на вимірюванні кількості електрики, яка витрачається на електрохімічну реакцію. В електрохімічній реакції бере участь або визначувана речовина (пряма кулонометрія), або утворюється речовина, яка кількісно реагує з визначуваною речовиною (кулонометричне титрування).
Основою кулонометрії є закони Фарадея. Згідно І-го закону Фарадея, маса речовини, що виділяється на електроді, прямо пропорційна кількості електрики, яка витратилась на електроліз:
m( A) kQ , |
(4.39) |
де m(A) - маса речовини A, г; Q - кількість електрики, Кл,
k - коефіцієнт, електрохімічний еквівалент речовини, г/Кл.
За ІІ-м законом Фарадея електрохімічний еквівалент речовини пропорційний її хімічному еквіваленту:
84
k 1 M ( A) ,
F n
де M(A) - молярна маса речовини, г/моль;
n - кількість електронів, які беруть участь в реакції; F - число Фарадея (F = 96500 Кл/моль).
Об’єднана формула законів Фарадея:
m( A) Q M ( A) .
Fn
(4.40)
(4.41)
Необхідні умови використання електрохімічної реакції в кулонометриному аналізі:
1) Електрохімічне перетворення речовини повинно перебігати із 100% виходом за струмом, тобто повинні бути відсутні побічні електродні реакції. При наявності побічних реакцій маса речовини, яка дійсно прореагувала на електроді, буде менша за масу речовини, розрахованої за формулою 4.39. Відношення цих мас називається виходом за струмом ( ):
|
m( A) Fn |
100% . |
(4.42) |
|
Q M ( A) |
||||
|
|
|
Побічні реакції відбуваються при наявності в розчині домішок, які окиснюються або відновлюються на електроді із значенням потенціалу при якому відбувається електроліз. Тому, для забезпечення 100% виходу за струмом необхідна відсутність речовин, які електроактивні при потенціалі електроду, або потенціал електроду повинен бути нижчим, ніж потенціал розкладу домішок.
2)Необхідно, щоб момент закінчення електрохімічної реакції можна було достатньо точно встановити.
3)Повинен існувати спосіб точного вимірювання кількості електрики, яка витрачається на електрохімічну реакцію.
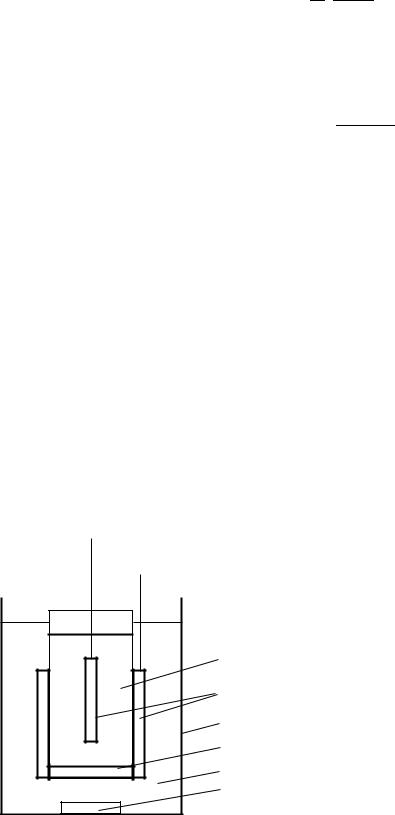
Кулонометричний аналіз проводиться на кулонометричній установці (рис. 4.12), яка складається з джерела постійного електричного струму (1), електролізера (2), прилада для вимірювання кількості електрики (3).
|
|
|
|
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
3 |
|
1 |
||
|
– |
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
6 |
Рис. 4.12. Принципова схема |
|||
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
8 |
кулонометричної установки. |
|
|
|
|
|
|
|
|
|
|
|
|
2 |
|
|
|
|
|
|
4 |
|
|
|
|
||||
|
|
|
5 |
|
||
7 |
|
|||||
Джерело електричного струму є або батареєю акумуляторів чи гальванічних елементів, або випрямлячем змінного струму з регульованою вихідною напругою чи струмом.
Електролізер складається з двох камер в яких знаходяться індиферентні електроди (8), зазвичай платинові. Камери з’єднуються пористою скляною перегородкою (4) або
85
електролітичним містком. Досліджувана речовина вміщається в робочу камеру (5) з робочим електродом, допоміжна камера (6) заповнюється розчином відповідного електроліта. В робочій камері розташована магнітна мішалка (7), робочий електрод має велику поверхню.
Якщо електроліз проводиться при постійній в часі силі струму (I, А) і можна точно виміряти час електролізу (t, с), кількість електрики визначається за формулою:
Q = It ( I = Const). (4.43)
Якщо сила струму під час електролізу змінюється, цією формулою користуватися не можна і кількість електрики визначити складніше:
t |
|
Q Idt (I = f(t)) |
(4.44) |
0 |
|
Для цього використовуються інтегратори струму або хімічні кулонометри. Це електролізери, у яких в умовах 100 % виходу за струмом утворюється певна речовина, точна кількість якої визначається або зважуванням, або визначенням об’єму газу, або титруванням. Переважно використовують водяний (Н2О) або йодидний (KJ) кулонометри.
4.4.2. Пряма кулонометрія.
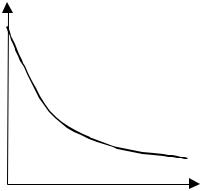
Пряма потенціостатична кулонометрія проводиться при постійному потенціалі робочого електрода. Склад електроліту робочої камери і потенціал робочого електроду вибираються такими, щоб забезпечити 100 % вихід за струмом. В цих умовах у процесі електролізу кількість носіїв заряду зменшується, а значить, зменшується електрична провідність розчину, що при постійному потенціалі призводить до зменшення сили струму
(рис. 4.13).
I
|
|
t |
|
Рис. 4.13. Зміна сили струму при потенціостатичній кулонометрії. |
|
||
При дифузійному режимі електролізу в робочій камері спадання струму описується |
|||
формулою: |
|
||
It = I0 e-Kt, |
(4.45) |
||
де I0 та It - сила струму на початку і в момент часу t; |
|
||
K - константа швидкості електролізу. |
|
||
Константа K залежить від розмірів робочої камери і властивості розчинника: |
|
||
K |
DS |
, |
(4.46) |
V
де D - коефіцієнт дифузії;
S - площа поверхні робочого електрода;
86

V - об'єм розчину в робочій камері;- товщина дифузійного шару.
При перемішуванні електроліту товщина дифузійного шару зменшується, що прискорює електроліз.
Теоретично електроліз завершується при безмежному часі електролізу, але, якщо зупинити електроліз при силі струму 0,01(I0) або 0,001(I0), буде забезпечена відносна точність результату – 1 або 0,1 %, відповідно.
Кількість електрики можна визначити з допомогою кулонометрів чи інтеграторів струму або розрахунковим шляхом, виходячи з формул 4.44 і 4.45:
|
I0 |
|
|
Q I0e Kt dt |
(4.47) |
||
K |
|||
0 |
|
||
|
|
K і I0 можна розрахувати з параметрів прямої лінії, побудованої в координатах lnIt - t. При наявності в розчині двох електроактивних речовин, потенціали розкладу яких
суттєво відрізняються, можливе їх послідовне визначення при різних потенціалах робочого електрода.
Незручність методу потенціостатичної кулонометрії полягає в порівняно складному визначенні кількості електрики.
Пряма амперостатична кулонометрія проводиться, коли джерело постійного струму забезпечує регульовану, постійну в часі силу струму не залежно від характеристики електролітичної комірки.
Амперостатична кулонометрія не використовується, якщо визначуваний іон знаходиться в розчині. У цьому випадку в процесі електролізу концентрація носіїв заряду зменшується, що призводить до зменшення електричної провідності і, при забезпеченні постійності сили струму, до збільшення потенціалу електрода (рис. 4.14).
U
t
Рис. 4.14. Зміна потенціалу при амперостатичній кулонометрії.
Зростання потенціала призводить до збільшення ймовірності побічних реакцій електролізу інших іонів розчину або розчинника. Внаслідок цього практично неможливо забезпечити 100 % вихід за струмом і одержати точні результати.
Якщо ж визначувана речовина знаходиться на електроді у твердому вигляді (наприклад, метал, оксиди металів), потенціал електроду практично не змінюється, тому що активнісь твердої фази не змінюється до повнго її зникнення. Після повного розчинення твердої речовини, яка вкриває електрод, потенціал електрода різко збільшується до величини, яка відповідає розчиненню іншої твердої фази, матеріалу електрода або потенціалу розкладу інших
87
іонів в розчині або розчинника. Цей стрибок потенціалу дозволяє зафіксувати час розчинення визначуваної речовини і розрахувати кількість електрики за формулою 4.43.
Амперостатична кулонометрія може використовуватися і для аналізу іонів в розчині, якщо вони попередньо електровідновлюються або окиснюються на електроді з утворенням твердої сполуки на поверхні електрода, з наступним розчиненням її при постійній силі струму. Цей метод застосовується для визначення товщини металічних покрить або оксидних плівок на різних поверхнях.
4.4.3. Кулонометричне титрування.
Кулонометричне титрування полягає в тому, що в розчин з речовиною, яку аналізують (A), додають допоміжний електроліт (Z), який при певних умовах піддається електролізу з утворенням проміжного реагента (T), який реагує з досліджуваною речовиною.
Z + e = T |
на електроді |
A + T = B |
в розчині |
Коли вся досліджувана речовина прореагує (точка еквівалентності), електроліз припиняють і визначають кількість електрики, яка пішла на електроліз за цей період. Особливістю кулонометричного титрування є відсутність розчину титранта. Речовина, яка вступає в реакцію титрування з визначуваною речовиною утворюється (генерується) в розчині на робочому електроді.
Кулонометричне титрування здійснюється, переважно, амперостатично, і кількість електрики визначається за формулою 4.43, де t - час, який пройшов від момента початку електролізу до момента, який відповідає точці еквівалентності. 100 % вихід за струмом забезпечують, беручи концентрацію допоміжного електроліта в 100 - 1000 разів більшу, ніж потрібно за стехіометрією реакції титрування. В результаті до точки еквівалентності концентрація електроліта практично не змінюється і не призводить до підвищення потенціалу електрода та проходження побічних реакцій, як в прямій амперостатичній кулонометрії.
Точка еквівалентності в кулонометричному титруванні може бути визначена або хімічним методом (з допомогою хімічних індикаторів), або фізико-хімічними методами (електрохімічними або фотометричними).
Визначувані речовини можуть бути як неелектроактивними, так і електроактивними.
У випадку електроактивних визначуваних речовин можливий наступний перебіг реакцій:
Z + e = T |
на електроді |
A + e = B |
на електроді |
A + T = B + Z |
в розчині |
У цьому випадку кількість електрики, яка проходить через електрод, витрачається на реакцію з визначуваною речовиною і допоміжним електролітом, але, внаслідок швидкого і повного проходження реакції в розчині (реакція титрування), сумарна кількість визначуваної речовини, що прореагувала, точно відповідає кількості електрики, яка проходить через електрод. Реакція в розчині призводить до регенерації допоміжного реактива (Z), концентрація якого до точки еквівалентності не змінюється і можна не давати його великого надлишку.
Як хімічні реакції між визначуваною речовиною і проміжним реагентом можуть використовуватися будь-які реакції, які використовуються в титруванні.
Приклади використання різних реакцій титрування Реакція осадження використовується для визначення іонів галогенів титруванням іонами
срібла електрогенерованими з срібного електрода: |
|
Hal- + Ag - e = AgHal↓ |
на аноді, |
88
Ag - e = Ag+ |
на аноді, |
|
Hal- + Ag+ = AgHal↓ |
в розчині. |
|
Точку еквівалентності зручно встановити потенціометрично |
за зміною потенціала |
|
срібного індикаторного електрода.
Реакція комплексоутворення зазвичай використовується з участю комплексону:
H2Y2- + Hg - 2e = HgY2- + 2H+ |
на аноді, |
Hg - 2e = Hg2+ |
на аноді, |
H2Y2- + Hg2+ = HgY2- + 2H+ |
в розчині. |
Точку еквівалентпості зручно визначити кондуктометрично. До точки еквівалентності електрична провідність різко збільшується через зростання в розчині рухливих іонів водню, після точки еквівалентності електропровідність збільшується повільніше через меншу рухливість іонів ртуті.
Реакція окиснення-відновлення. Для визначення Ce4+ у розчин додають Fe3+, на платиновому катоді відбуваються реакції:
Точка електродом.
Реакція кислотно-основної взаємодї. У водних розчинах допоміжною речовиною є розчинник - вода, яка при електролізі на катоді або на аноді генерує відповідно H+ та OH-.
Наприклад, на катоді:
2H+ +2e = H2 ↑
2H2O + 2e = H2↑ + 2OH-
з відповідною реакцією в розчині:
H+ + OH- = H2O
Точку еквівалентності можна визначити з допомогою відповідних кислотно-основних індикаторів або потенціометрично зі скляним індикаторним електродом.
Визначеня різних неелектроактивних органічних сполук (оксинів, амінів, фенолів,
нафтолів, хінолінів) можна здійснити електрогенерованим Br2 з броміду на Pt аноді:
2Br- - 2e = Br2,
з наступною реакцією в розчині:
C6H5OH +3Br2 = C6H2Br3OH + 3Br- + 3H+
Точку еквівалентності визначають потенціометрично або йодометрично.
Масу визначуваних речовин розраховуюь за формулою 4.39, де n - кількість електронів, яка еквівалентна молекулі визначуваної речовини з урахуванням стехіометрії реакцій, які відбуваються на електроді і в розчині. В останньому прикладі f(C6H5OH) = 1/6.
Кулонометричне титрування більш чутливе ніж інші титриметричні методи і при визначенні малих кількостей речовини суттєвою стає систематична індикаторна помилка фіксування точки еквівалентності. Тому, крім визначення часу електролізу до кінцевої точки титрування, проводять холостий дослід, який здійснюють в тих самих умовах тільки без досліджуваної речовини. Для одержання точних результатів від часу електролізу визначуваної речовини віднімають час фіксування точки еквівалентності холостого досліду.
Переваги кулометричноо титування:
1. Виключається необхідність приготування і стандартизації розчину титранту.
89
2.Можливість використання для титрування речовин, стандартні розчини яких нестабільні (Cl2, Br2, Ag+, Cu2+, Ti3+, Sn2+ та інші).
3.Розчин в процесі титрування не розбавляється.
4.В одному і тому ж розчині допоміжного електроліту можливе проведення титрування декількох зразків.
5.Титування може бути повністю автоматизоване.
6.Можна визначати в одному розчині кілька речовин.
Питання для самоконтролю.
20.На чому грунтуються кондуктометричні методи аналізу?
21.Електрична провідність розчинів електролітів – питома, еквівалентна, рухливість.
22.Принципова схема кондуктометричної установки.
23.Прямий кондуктометричний аналіз, його переваги і недоліки.
24.Кондуктометричне титрування, його можливості.
25.Закон Фарадея.
26.Вихід за струмом.
27.Потенціостатична кулонометрія.
28.Амперостатична кулонометрія.
29.Кулонометричне титрування.
5. РАДІОМЕТРИЧНІ МЕТОДИ АНАЛІЗУ
5.1. Загальна характеристика радіометричних методів аналізу. Поняття про природні та штучні радіоізотопи (радіонукліди)
Радіометричні методи аналізу грунтуються на вимірюванні інтенсивності радіоактивного випромінювання (аналітичний сигнал), яке виникає при самочинному розпаді ядер радіоактивних ізотопів (радіонуклідів).
Радіонукліди — це сукупність атомів з однаковим зарядом ядра Z (кількість протонів у ядрі) і з різним масовим числом А (кількість протонів та нейтронів), які здатні самочинно розпадатись із виділенням ядерної енерґії у вигляді радіоактивного випромінювання.
Радіонукліди характеризуються нестійкою конфіґурацією ядерних систем, внаслідок чого в ядрах радіонуклідів відбуваються самочинні перетворення, які призводять до вивільнення надлишкової енерґії у вигляді радіоактивного випромінювання і переходу ядер із нестійкого у стабільний стан.
В даний час відомо ~ 2000 радіонуклідів, серед яких лише ~ 300 зустрічаються в природі. Природні — це радіонукліди, які утворилися і постійно знову утворюються без участі
людини. Це всі ізотопи елементів із Z 84 (полоній Po), які складають сімейства:
1) |
торію 23290 Th … 20882 Pbстабільний, |
||
2) |
актино-урану |
23592 U … |
20782 Pbстабільний, |
3) |
урано-радію |
23892 U … |
20682 Pbстабільний, |
а також ізотопи більш легких елементів, генетично не зв’язаних між собою:
13857 La, 14762 Sm, 18775 Re, …
90
Штучні радіонукліди одержують в ядерних реакторах і прискорюючих установках в результаті ядерних реакцій, бомбардуючи ядра атомів стабільних або радіоактивних ізотопів пучками легких елементарних частинок (α-частинками, нейтронами, протонами) або γ- квантами.
Наприклад, при бомбардуванні ядер 2713 Al можливі такі ядерні реакції, внаслідок чого одержують штучні нукліди P і Na:
2713 Al + 42 α = 3015 P + 01 n
2713 Al + 01 n = 2411 Na + 42 α
5.2. Типи радіоактивних розпадів та їх характеристика.
Самочинні перетворення, які відбуваються в ядрах радіонуклідів, можна розділити на декілька типів, основними із яких є:
α-розпад, β-перетворення, спонтанний поділ та інші. Ці перетворення супроводжуються виділенням радіоактивного випромінювання.
Розглянемо кожний із цих типів перетворень зокрема і поряд з цим подамо коротку характеристику кожного виду радіоактивного випромінювання.
5.2.1. α-Розпад та характеристика α-випромінювання.
α-Розпад — це розпад ядра із виділенням α-частинки. Він характерний для важких радіонуклідів (Z > 78).
α-Частинка — це ядро атома гелію, яка складається з двох протонів і двох нейтронів
4 2
α
Випромінюючи α-частинку ядро радіонукліду перетворюється в ядро нового елемента, заряд якого буде меншим на дві, а масове число — на чотири одиниці, за схемою:
AZ X
4 2
α + AZ 42 Y.
Вихідний елемент перетворюється на елемент, розташований на 2 клітинки ліворуч у періодичній таблиці.
Наприклад, 23290 Th
4 2
α +
22888 Ra.
Кожен вид радіоактивного випромінювання характеризується: енерґією, швидкістю поширення, проникною та йонізаційною здатністю.
α-Випромінювання — це потік позитивно заряджених α-частинок (ядер атомів гелію), які рухаються із швидкістю ~ 20000 км/с і володіють енерґією від 2 до 9 МеВ.
Порівняно з іншими видами випромінювання, α-випромінювання характеризується найбільшою йонізаційною та найменшою проникною здатністю; α-частинки витрачають практично всю свою енерґію на йонізацію середовища, пролітаючи в повітрі від 2 до 20 см залежно від енерґії; вони затримуються шаром алюмінію товщиною 0,01–0,03 мм чи аркушем паперу.
5.2.2. β - Розпад та характеристика β - випромінювання.
β-Розпад — це самочинне взаємне перетворення нейтронів і протонів всередині ядра, яке включає β–-розпад, β+-розпад та електронне захоплення (ЕЗ).
β–-розпад (електронний розпад) — це найбільш поширений вид розпаду, який характерний для нуклідів із надлишком нейтронів.
91
Суть його полягає в тому, що один із нейтронів в середині ядра перетворюється в протон і випромінює β–- частинку (електрон) та антинейтрино ν:
|
1 n |
1 p + β– + ν |
|
|
0 |
1 |
|
При цьому виді розпаду утворюється ядро елемента із тим же масовим числом, але із |
|||
зарядом на одну одиницю більшим, за схемою: |
|
|
|
|
A X β– + |
A Y + ν. |
|
|
Z |
|
Z 1 |
Вихідний елемент перетворюється на елемент, розташований на 1 клітинку праворуч у |
|||
періодичній таблиці. |
|
|
|
Наприклад, 40 K β– + |
40 Ca + ν |
|
|
19 |
20 |
|
|
Цей тип розпаду супроводжується β–-випромінюванням. β–-Випромінювання — це потік |
|||
негативно заряджених β–-частинок (електронів), |
які рухаються із швидкістю від 2.105 до |
||
3.105 км/с та володіють енерґією від 0,5 до 1 МеВ.
β–-частинка — це елементарна частинка з масою та зарядом електрона; антинейтрино ν — це частинка без заряду і з масою близькою до нуля.
Йонізаційна здатність β–-частинок у порівнянні з α-частинками майже в 100 разів менша, що пов’язано з меншими значеннями їх енерґії, маси, розмірів та заряду. Зате їх проникна здатність більша. Шлях пробігу β–-частинок у повітрі складає від кількох сантиметрів до кількох метрів, залежно від енерґії, а в металі (залізо) — до 1,5 мм.
β+-розпад (позитронний розпад) зустрічається рідко і є характерним переважно для штучно одержаних радіонуклідів із надлишком протонів.
Суть його полягає в тому, що один із протонів ядра перетворюється в нейтрон, випромінюючи β+-частинку (позитрон) і нейтрино ν:
11 p 01 n + β+ + ν
При цьому виді розпаду утворюється ядро елемента з тим же масовим числом, але із зарядом на одну одиницю меншим, за схемою:
AZ X β+ + ZA-1 Y + ν.
Вихідний елемент перетворюється на елемент, розташований на 1 клітинку ліворуч у періодичній таблиці.
Наприклад, 2211 Na β+ + 2210 Ne + ν.
Позитрон характеризується малим часом життя, оскільки він миттєво взаємодіє з електроном оболонки власного або сусіднього атома, перетворюючись у два γ-кванти з енерґією 0,51 МеВ кожен (явище аннігіляції):
β+ + e– 2γ.
Електронне захоплення (К-захоплення) – тип ядерного перетворення, протилежний до β+- розпаду, хоча він також, як і позитронний β+-розпад, характерний для протононадлишкових ядер, переважно важких радіонуклідів і зустрічається рідко.
Суть цього ядерного перетворення полягає в тому, що один із протонів ядра захоплює електрон, як правило, з найближчого К-енерґетичного рівня, перетворюючись в нейтрон. В результаті ЕЗ проходить перебудова електронних оболонок (перехід e– з L-орбіталі на місце, що звільнилося на К-орбіталі, і т.д.), тому процес супроводжується випромінюванням жорстких рентґенівських квантів hν:
11 p + e– 01 n + hν
92