

Konspekt_lekcii
.pdfселективний до титранта, бо з його допомогою можна визначати цілий клас речовин, які реагують з цим титрантом.
Для потенціометричного титрування можуть використовуватися всі реакції, які задовольняють вимогам, які ставляться до реакцій титрування, крім вимоги наявності індикатора.
1. Кислотно-основне титрування. Як правило, для такого титрування беруть скляний індикаторний електрод, хлоридно-срібний електрод порівняння і вимірюють pH розчину з допомогою pH-метра.
Потенціал індикаторного електрода буде залежати від рН розчину:
E = 0,059lg[H+] = -0,059ּlg(pH).
При титруванні сильної кислоти (Vk0 = 100 мл, Сk0 = 0,1 M) сильною основою ( Сосн0 = 0,1 M) потенціал до точки еквівалентності обчислюють за формулою:
|
C 0V 0 |
C 0 V 0 |
0,1(100 V |
) |
|
|
|
E 0,059 lg |
k k |
осн осн |
0,059 lg |
осн |
|
, |
(4.10) |
|
|
|
|
||||
1 |
V 0 |
V |
100 V |
|
|
|
|
|
|
|
|
||||
|
k |
осн |
осн |
|
|
|
|
Потенціал після точки еквівалентності визначається за рівнянням:
|
0,059 lg |
|
|
10 14 |
|
0,059 lg |
10 14 (V |
|
100) |
|
|
||
E2 |
|
|
|
|
|
|
осн |
|
, |
(4.11) |
|||
|
V |
|
С 0V 0 |
|
0,1(V |
100) |
|||||||
|
|
|
C 0 |
|
|
|
|
|
|||||
|
|
|
осн осн |
к k |
|
|
осн |
|
|
|
|
||
|
|
|
|
V |
|
V 0 |
|
|
|
|
|
|
|
|
|
|
|
осн |
|
k |
|
|
|
|
|
|
|
Стрибок потенціала Е = Е1 – Е2 |
буде тим більший, чим більші початкові концентрації |
||||||||||||
кислоти і основи. При необхідній точності визначення об’єму точки еквівалентності – 0,1%, стрибок спостерігається при концентраціях Ck0 і Cосн0 більших, ніж 10-4 М.
Складніше визначити стрибок потенціала при титруванні слабких кислот або основ. Розглянемо титрування слабкої кислоти сильною основою. Концентрацію іонів водню і рН до початку титрування розраховують за відомими формулами:
[H+] |
Kc Ck |
|
|
|
||||||
pH |
1 |
pK |
|
|
1 |
lg C |
|
|
||
|
k |
|
k |
|
||||||
2 |
|
|
|
2 |
|
|
|
|||
|
|
|
|
|
|
|
|
|||
Тоді потенціал в початковій точці буде рівний: |
|
|
||||||||
E 0,0295( pKk |
lg Ck ) . |
(4.12) |
||||||||
В процесі титрування в розчині утворюється буферна суміш, яка складається із залишку слабкої кислоти та її солі. Для такої суміші рН і, відповідно, Е обчислюють за формулами:
pH pK lg |
Ck |
|
|
Cc |
|
||
E 0,059( pK lg Cc lg Ck ) . |
(4.13) |
||
В точці еквівалентності в розчині присутня сіль слабкої кислоти та сильної основи, яка гідролізує, і рН та Е розраховують за рівняннями:
pH 7 |
1 |
pKk |
1 |
lg Cc |
|
|||||
2 |
2 |
|
||||||||
|
|
|
|
|
|
|
|
|||
E 0,059(7 |
1 |
pKk |
1 |
lg Cc ) , |
(4.14) |
|||||
2 |
2 |
|||||||||
|
|
|
|
|
|
|
|
|||
після точки еквівалентності при додаванні сильної основи потенціал визначають так само, як і при титруванні сильної кислоти (4.11).
63

Стрибок потенціала в даному титруванні буде тим більший, чим більша константа іонізації кислоти та її концентрація. На рис. 4.2 приведені приведені криві потенціометричного титрування кислот різної сили сильною основою.
pH |
|
|
|
Рис. 4.2 |
Криві потенціометричного титрування. |
|
1 |
– сильна кислота, |
|
2 |
– слабка кислота, |
|
3 |
– суміш сильної та слабкої кислот. |
V1т.е |
V2т.е Vлугу, мл |
|
Як видно з рисунка, слабких кислот або основ стрибок титрування спостерігається, коли
К ≥ 10-8, а рК ≤ 8.
При титруванні суміші кислот або основ різної сили при різниці силових показниківpK > 4 спостерігається два стрибки титрування. У цьому випадку можливе визначення двох компонентів в одному розчині без розділення.
2. Окисно-відновне титрування. Для такого титрування як індикаторний використовують переважно платиновий електрод. Це платинова дротинка або платиноване скло. Електрод порівняння може бути будь-який.
Стрибок титрування залежить від стандартних окисно-відновних потенціалів визначуваної речовини і титранту. При необхідній точності 0,1% стрибок титрування буде при умові:
|
|
|
E 0 |
E 0 |
|
|
1 |
|
1 |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
0,177 |
|
|
|
, |
(4.15) |
||
|
|
|
|
|
|
||||||
|
|
|
1 |
2 |
|
|
|
|
|
|
|
де E 0 |
і E 0 |
|
|
|
|
n1 |
|
n2 |
|
|
|
|
|
|
|
|
|
||||||
- стандартні окисно-відновні потенціали визначува ної речовині і титранта; |
|||||||||||
1 |
2 |
|
|
|
|
|
|
|
|
|
|
n1 і n2 - кількості електронів, які беруть участь у півреакціях окиснення і відновлення. |
|||||||||||
При n1= n2 |
крива титрування симетрична відносно точки еквівалентності. Якщо півреакції |
||||||||||
окиснення або відновлення відбуваються зі зміною кількості моль, стрибок титрування залежить і від концентрації.
Прикладом окисно-відновного потенціометричного титрування може бути визначення
іонів Fe2+ за допомогою дихромату калію в кислому середовищі: |
|
||
6Fe2+ + Cr2O72- + 14H+ → 6Fe3+ + 2Cr3+ + 7H2O |
|
||
Fe2+ -1e → Fe3+ |
6 |
|
6 |
Cr2O72- + 14H+ + 6е-→2Cr3+ + 7H2O |
|
1 |
|
Стандартні потенціали окисно-відновних пар рівні:
E 0 |
|
|
1,33 B; E 0 |
0,77 B. |
Cr O2 14H |
|
Fe3 |
|
|
2 |
7 |
3 |
|
Fe2 |
|
2Cr |
|
||
|
|
|
|
|
До точки еквівалентності потенціал обчислюють за рівнянням Нернста для редокс-пари
Fe3 Fe2 :
64

E |
|
|
E 0 |
|
0,059 lg |
[Fe3 ] |
(4.16) |
Fe3 |
|
|
|
||||
|
2 |
Fe3 |
Fe2 |
[Fe2 ] |
|
||
|
|
Fe |
|
|
|
|
|
В точці еквівалентності в розчині присутні в основному йони Fe2+ і Cr3+; в невеликих кількостях присутні також йони Fe2+ і Cr2O72-, так як в точці еквівалентності існує рівновага
системи Fe3 Fe2 і Cr2O72 14H 2Cr3 . Тому потенціал в цей момент титрування розраховують за рівнянням:
|
E 0 |
6E 0 |
|
|
|
Fe3 |
Cr O2 14H |
|
|
|
Fe2 |
2 |
7 |
3 |
Eт.е. |
|
2Cr |
||
|
|
|
||
|
1 6 |
|
|
|
|
|
|
|
|
0,77 6 1,33 1,25 В 7
Після точки еквівалентності в розчині зростає надлишок K2Cr2O7 при практично незмінній концентрації Cr3+ (розведення розчину титрантом, як правило, не враховується!), тому потенціал системи зручно обчислювати за формулою:
E |
Cr2O72 14H |
E 0 |
|
0,059 |
lg |
[Cr2O7 ][H ]14 |
|
[Cr 3 ]2 |
|||||
|
Cr2O72 14H |
2Cr3 |
6 |
|
||
|
2Cr3 |
|
|
|
|
Для окремого визначення суміші двох відновників або окисників необхідно, щоб різниця їх стандартних окисновідновних потенціалів була більше 0,36 В.
3. Титриметричний метод осадження. Найбільш поширеним методом осаджувального титрувння є аргентометрія, в якому титрантом є нітрат срібла. З його допомогою можна визначати іони Cl-, Br-, I-, SCN- та ін., які утворюють з іонами срібла малорозчинні сполуки. Використовується срібний індикаторний електрод і каломельний електрод порівняння, який з'єднується з досліджуваним розчином сольовим містком з KNO3.
Застосування методу осадження в потенціометричному титруванні розглянемо на прикладі визначення срібла хлорид-йонами.
Потенціал срібного електрода до точки еквівалентності визначають за формулою:
E E 0 |
0,059 lg[ Ag ] |
Ag |
|
|
Ag0 |
В точці еквівалентності всі йони срібла будуть переведені в осад AgCl. Концентрація йонів срібла у цьому випадку визначається розчинністю осаду і може бути обчислена з добутку розчинності:
[Ag+] = [Cl-] = 
 ДРAgCl
ДРAgCl
Потенціал в точці еквівалентності буде мати значення:
E E 0 |
|
|
|
|
|
0,059 lg ДР |
AgCl |
||
Ag |
|
|
||
|
|
|
|
|
|
|
Ag0 |
|
|
Після точки еквівалентності концентрація йонів срібла залежить від концентрації надлишку хлорид-йонів і знову буде визначатись добутком розчинності AgCl:
[ Ag ] |
ДРAgCl |
|
[Cl ] |
||
|
Потенціал срібного індикаторного електрода після точки еквівалентності розраховують за формулою:
65
E E 0 |
|
0,059 lg |
ДРAgCl |
||
|
|
|
|
||
Ag |
|
|
[Cl |
] |
|
|
Ag0 |
|
|
||
|
|
|
|
|
|
На кривій залежності потенціала від об’єму титранта спостерігається стрибок потенціала, який відповідає точці еквівалентності. Стрибок потенціалу залежить від концентрації реагентів
ідобутку розчинності одержаного осаду. Стрибок буде тим більший, чим більша концентрація
іменший ДР. При точності титрування 0,1 % стрибок титрування буде при умові pДР + 2lgC>6.
При наявності в розчині декількох речовин, які дають малорозчинні речовини з одним титрантом, декілька стрибків титрування спостерігатиметься, якщо відношення добутків розчинності сполук перевищуватиме 104. Наприклад, суміш галогенідів (Cl- і J-) може бути відтитрована без розділення нітратом срібла (рис. 4.3).
E
V1т.е |
V2т.е V (AgNO3), мл |
Рис. 4.3. Крива потенціометричного титрування суміші галогенідів (Cl- і J-) розчином AgNO3.
Срібний електрод зафіксує два стрибки в ході титрування. Перший стрибок свідчить про відтитровування йодид-йона (ДРAgJ = 8,3. 10-17), може бути використаний для розрахунку його вмісту. Другий стрибок відноситься до закінчення осадження хлорид-йонів (ДРAgCl = 1,78. 10- 10). За другим стрибком можна розрахувати сумарний вміст галогенідів або концентрацію хлорид-йона, якщо концентрація J--йонів буде відома з даних титрування за першим стрибком.
4. Комплексометричне титрування. Титрантом в комплексонометричному титруванні є розчин ЕДТА, який утворює з багатозарядними йонами малодисоційовані внутрішньокомплексні сполуки. В процесі титрування спостерігається стрибок рМе, бо як індикаторні електроди використовують різні катіон-селективні електроди. Якщо в розчині присутні два йони, які утворюють з ЕДТА комплекси з великою різницею констант стійкості, можливе фіксування двох стрибків титрування і визначення двох катіонів.
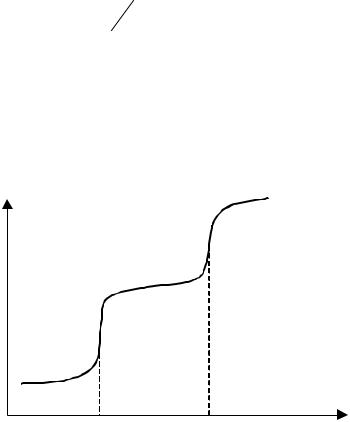
Для точнішого фіксування точки еквівалентності потенціометричного титрування будують криві титрування в координатах dE/dV - V або d2E/dV2 – V (рис. 4.4). У першому випадку Vт.е. відповідає максимуму кривої, у другому - перетину кривої з віссю V.
66
Е
а
ΔE |
Vт |
|
ΔVт |
|
|
|
|
|
d2E |
Vт |
dVт |
|
Vт
Рис. 4.4. Визначення точки еквівалентності за кривою титрування (а), її першою похідною (б), другою похідною (в).
Розроблені автоматичні титратори, в яких титрант подається з постійною швидкістю і будується залежність першої або другої похідної сигналу потенціометра від часу. Подача титранта автоматично припиняється при досягненні максимума першої похідної сигналу або 0 другої похідної. При апаратному диференціюванні сигналу потенціометра можливе фіксування точки еквівалентності не тільки при наявності стрибка титрування, а й при наявності тільки перегину на кривій титрування.
Переваги потенціометричного титрування:
1.Можливість аналізу забарвлених або каламутних розчинів.
2.Визначення концентрацій декількох речовин в розчині.
3.Можливе титрування не тільки у водних, але і в неводних або змішаних розчинах.
4.Вплив різних факторів на реальний потенціал індикаторного електрода практично не впливає на результат титрування. Точна настройка шкали прилада на істинну величину потенціала не обов'язкова.
Питання для самоконтролю.
1.Класифікація електрохімічних методів аналізу.
2.Основи потенціометричних методів аналізу.
3.Класифікація електродів. Електроди I, II, III роду, індиферентні та іонообмінні.
4.Аналітичний сигнал потенціометричних методів аналізу.
5.Вимоги до електродів.
6.Індикаторні та стандартні електроди.
7.Вимірювання потенціалів в потенціометрії.
8.Пряма потенціометрія. Переваги.
9.Потенціометричне титрування, його можливості.
10.Потенціометричне титрування, основане на реакціях нейтралізації, окисненнявідновлення, осадження, комплексонометрії.
67
4.2. Полярографічні методи аналізу
4.2.1. Види поляризації.
Полярографічні методи аналізу грунтуються на явищах поляризації на електроді з малою поверхнею, які виникають при проходженні електричного струму через розчин електроліту.
Цей метод запропонований чеським вченим Я. Гейровським у 1922 році і детально розроблений ним та його учнями. У 1959 році за ці роботи Я. Гейровський удостоєний Нобелевської премії в галузі хімії.
Поляризацією називається явище, зумовлене проходженням електричного струму через розчин електроліту, яке викликає відхилення потенціалу електрода від рівноважного значення,
розрахованого за рівнянням Нернста. Поляризація буває хімічна, електрохімічна і концентраційна.
Якщо в розчин електроліту занурити 2 індиферентних електрода (наприклад, платинові) і сполучити їх з зовнішнім джерелом постійного електричного струму, то на одному з електродів (катоді) починається процес відновлення катіонів, на іншому (аноді) - окиснення аніонів. Через розчин буде проходити електричний струм, а на електродах будуть відкладатися продукти електродних реакцій. Наприклад, якщо у воді розчинена сіль CdCl2, катод вкриється металічним Cd, а анод - абсорбованим Cl2. Через деякий час індиферентні електроди перетворюються в електроди І роду: Cd│Cd2+ i Cl2│Cl–, які складають гальванічний елемент з ЕРС = EoCl2 /Cl- – EoCd2+/Cd = 1,36 - (-0,40) = 1,76 В при нормальних умовах, спрямованою проти прикладеного джерела струму. Якщо напруга зовнішнього джерела струму менша ЕРС утвореного гальванічного елемента, через деякий час струм припиняється.
Процес виникнення різниці потенціалів між зануреними в розчин індиферентними електродами під впливом електролізу називається хімічною поляризацією. ЕРС, яка при цьому виникає, називають ЕРС хімічної поляризації (Ехп). Ехп залежить від катіонного і аніонного складу електроліта і його концентрації.
Очевидно, що для забезпечення перебігу електролізу, до електродів необхідно прикласти напругу більшу ніж Ехп. Додаткова різниця потенціалів, необхідна для забезпечення перебігу електролізу з певною швидкістю називається електрохімічною поляризацією або перенапругою.
Перенапруга ( ) залежить від властивості іона і матеріала електрода та характера його поверхні. Потенціал, при якому починається електроліз називається потенціалом розкладу (Ер).
Ер = Ехп + .
В результаті електролізу простір навколо електрода збіднюється відповідними іонами. Виникає різниця конценрацій іонів в основній масі розчину (Со) і в приелектродному шарі (Сs). Цей градієнт концентрацій викликає додаткову різницю потенціалів.
Додаткова різниця потенціалів, яка виникає внаслідок збіднення приелектродного шару іонами, називається концентраційною поляризацією. Її можна обчислити за рівнянням:
E |
кп |
|
0,059 |
lg |
C0 |
, |
(4.17) |
|
n |
Cs |
|||||||
|
|
|
|
|
||||
|
|
|
|
|
|
4.2.2. Принципова схема полярографічної установки.
Полярографічна установка (рис. 4.5) складається з електролітичної комірки, джерела постійного електричного струму, приладів регулювання напруги, вимірювання напруги і сили струму.
68

Рис. 4.5. Принципова схема полярографічної установки.
Електролітична комірка (електролізер) складається з двох електродів, занурених в розчин, який аналізують. На одному з електродів (робочому) відбувається окиснення або відновлення визначуваної речовини, другий електрод є електродом порівняння.
Електродом порівняння може бути донна ртуть, яка збирається на дні електролітичної комірки від ртутного краплинного електрода (РКЕ), або будь-який стандартний електрод. Часто використовується нормальний або насичений каломелевий електрод (НКЕ), який з'єднується з досліджуваним розчином солевим електролітичним містком.
Перевагою донної ртуті як електрода порівняння є те, що амальгама, яка збирається на дні полярографічної комірки, має велику поверхню, і біля неї густина струму мала, що не викликає концентраційної поляризації. Крім цього відбувається процес анодного розчинення визначуваного металу з амальгами в розчин. При цьому концентрація визначуваних йонів у розчині під час аналізу не змінюється.
Джерелом постійного струму є батарея акумуляторів (Ак) або стабілізований випрямляч змінного струму, які забезпечують напругу на електродах 2-3 В. Джерело струму з'єднане з реохордом (Р), рухомий контакт якого дозволяє плавно змінювати напругу на електродах, яка вимірюється вольтметром (V). Сила струму електролізу вимірюється гальванометром (Г).
Оскільки концентраційна поляризація безпосередньо пов'язана з концентрацією електроактивних іонів (деполяризатора), вона є головна для аналізу. Потенціал концентраційної поляризації залежить від градієнту концентрації, який тим більший, чим більша густина струму біля поверхні електроду. Тому площа поверхні робочого електроду повинна бути меншою ніж поверхня електроду порівняння в 100-1000 разів.
Гейровський використовував для катодної поляризації ртутний краплинний електрод, тобто поверхню ртуті, яка витікає через скляний капіляр занурений в досліджуваний розчин. Для анодної поляризації використовують індиферентні електроди (прерважно Pt) з малою поверхнею (діаметром менше 1 мм).
Переваги ртутного краплинного електрода для катодної поляризації:
1. Ртуть, яка витікає в розчин із скляного капіляра, утворює маленькі краплі, що забезпечує велику густину струму навіть при силі струму декілька мікроампер.
69
2.На поверхні ртуті йони водню відновлюються з великою перенапругою, що дозволяє полярографувати йони металів, які знаходяться в ряду напруги лівіше водню, аж до йонів лужноземельних і лужних металів. Ртуть можна використовувати при потенціалах +0,3...-2,0 В.
3.Більшість металів, які відновлюються на поверхні краплі, утворюють амальгами і дифундують вглиб краплі. Активність метала при цьому на поверхні краплі зменшується, що зменшує потенціал хімічної поляризації. Крім того, після скапування краплини з розчиненим металом, утворюється нова краплина чистої ртуті. Таким чином, поверхня електрода постійно оновлюється і усувається хімічна поляризація електрода.
4.2.3. Полярографічна хвиля.
Розглянемо, як буде змінюватися струм, який проходить через електролітичну комірку, із зростанням напруги прикладеної до електродів. Якщо визначувана речовина має потенціал розкладу Ер, то при збільшенні потенціалу електрода від 0 до Ер струм не повинний проходити. Насправді на цій ділянці (АВ рис. 4.6) проходить невеличкий струм, який називається залишковим (Ізал) .
I, мкА |
|
D |
|
) |
Рис. 4.6. Катодна вольтамперна |
|
крива (полярографічна хвиля). |
Е1/2 |
Е, В |
Цей струм має два складники: конденсаторний (Іс) та фарадеєвський (Іf):
Із = Іс + Іf |
(4.18). |
Причиною виникнення конденсаторного струму є те, |
що при збільшенні потенціалу |
електрода біля нього збираються йони протилежного заряду, потенціал розкладу яких більш негативний ніж потенціал електрода, і утворюють подвійний електричний шар за типом конденсатора. Фарадеєвський струм виникає внаслідок електровідновлення незначної кількості домішок, які присутні в розчині.
Залишковий струм має величину порядку 10-7 А, що обмежує чутливість метода до 10-5 - 10-7 моль/л визначуваної речовини.
Збільшення напруги на електродах, яке призводить до потенціалу електрода більш негативного, ніж Ер викликає процес відновлення аналізованої речовини на краплині ртуті, внаслідок чого струм у колі різко зростає (ділянка ВС). Рушійною силою струму електролізу є дифузія йонів за рахунок градієнту концентрацій визначуваних йонів у розчині і поблизу поверхні електроду (Iд) і міграція йонів за рахунок електростатичного притягання зарядів йону і поверхні електрода (Ім). Сила струму електролізу (Іе) дорівнює:
70
Iе = Iд + Iм |
(4.19). |
Дифузійна складова (Iд ) визначається за формулою: |
|
Iд = FDSn(Co - Cs)/ , |
(4.20) |
де F - число Фарадея,
D - коефіцієнт дифузії,
S - площа поверхні електрода,
n - число електронів, яке бере участь в електродній реакції,- товщина дифузійного шару.
Оскільки концентрація іонів у розчині (Со) залишається практично незмінною, збільшення сили дифузійного струму може здійснюватися за рахунок зменшення концентрації йонів у приелектродному шарі (Сs). Таким чином, сила дифузійного струму може збільшуватися до свого максимального значення Iгр = FDSnCo/ при Сs = 0 і не збільшується при подальшому збільшенні потенціалу електрода (ділянка CD).
Сила міграційного струму (Iм) залежить від заряду іонів, потенціалу електрода, в'язкості розчину і температури. Міграційний струм утруднює інтерпретацію полярограм, тому його усувають додаючи у розчин надлишок йонів, які відновлюються при більш негативному потенціалі, ніж визначуваний йон. Ці йони вводять у розчин у вигляді фонових електролітів, які є переважно солями одновалентних катіонів: KCl, LiF, NaNO3, NH4Cl; розчинами лугів і кислот, а також метанол, ацетонітрил та інші. Йони фону екранують поверхню електрода, зменшуючи його ефективний заряд. Якщо концентрація іонів фону в 100-1000 разів перевищує концентрацію визначуваних іонів, міграційний струм практично зменшується до нуля, і струм електролізу дорівнює дифузійному струму (Iе = Iд).
Залежність сили дифузійного струму від потенціалу електрода, зображена на рис. 4.6, називається вольтамперною характристикою або полярограмою, а ділянка BC називається полярографічною хвилею. Полярограма є аналітичним сигналом полярографічних методів аналізу.
Прилади, які дозволяють фіксувати полярограму, називаються полярографами. У перших полярографів зміна потенціала електродів проводилася вручну і після такої зміни записувався струм електролізу. За цими даними будувалася полярограма. У сучасних електронних автоматичних полярографах всі операції виконуються автоматично із записом полярограми на рухомій стрічці самописця або на дисплеї монітора ЕОМ.
4.2.4. Якісний полярографічний аналіз.
Якісний полярографічний аналіз грунтується на вимірюванні потенціалу півхвилі йону,
який відновлюється. Потенціалом півхвилі називається потенціал, при якому досягається сила струму, яка дорівнює половині граничного дифузійного струму (рис. 4.6). Потенціал півхвилі позначається - Е1/2 і залежить від природи іону, який відновлюється, природи і концентрації фонового електроліта, pH розчину і не залежить від концентрації йону. Потенціали півхвиль різних неорганічних і органічних речовин у фонових електролітах різного складу виміряні експериментально і наводяться в довідниковій літературі.
Процедура якісного аналізу полягає у фіксуванні полярограми досліджуваного розчину у певному фоновому електроліті, вимірюванні потенціалу півхвилі і пошуку у довідникових даних речовини, потенціал півхвилі якої у цьому ж фоні дорівнює виміряній величині потенціалу півхвилі.
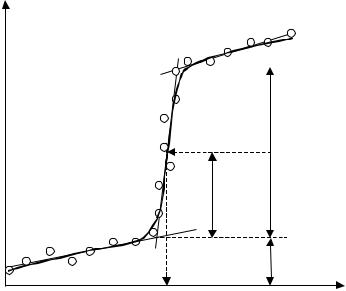
Потенціал півхвилі можна виміряти графічно, провівши дотичні прямі через експериментальні точки дифузійного струму в межах полярографічної хвилі, а також до і після
71
хвилі. Дві точки перетину цих прямих відповідають граничному дифузійному струму. Відрахувавши потенціал, який відповідає половині відстані між точками перетину, вимірюють потенціал півхвилі визначуваного іону.
Точніше визначити потенціал півхвилі можна з допомогою рівняння полярографічної хвилі для оборотних процесів Гейровського-Ільковича:
E E 12 |
|
0,059 |
lg |
I |
, |
(4.21) |
|
|
|||||
n |
I гр I |
де I - сила струму у будь-якій точці полярографічної хвилі.
Якщо побудувати графік залежності lgI/(Iгр - I) від Е, то одержимо пряму лінію, яка перетинає вісь абсцис (рис. 4.7).
lgI/(Iгр - I)
α
Е½ |
Е |
Рис. 4.7. Вольтамперна крива в напівлогарифмічних координатах
Якщо І = Iгр/2, то lgI/(Iгр - І) = 0 і Е = E1/2 . Тому координата точки перетину відповідає потенціалу півхвилі. З кута нахилу прямої можна визначити кількість електронів, яка бере участь в електродній реакції, n = 0,059tg .
При наявності в розчині декількох йонів можлива окрема ідентифікація, якщо їх потенціали півхвилі відрізняються не менше ніж на 0,2 В. Якщо потенціали півхвилі в одному фоні відрізняються недостатньо, для окремої ідентифікації можна замінити фоновий електроліт. Наприклад, у 1М KCl E1/2(Ni2+) = -1,1 В; E1/2(Zn2+) = -1,02 В, натомість у 1М NH3 + 0,2 M NH4Cl – E1/2(Ni2+) = -1,06 В, а E1/2(Zn2+) = -1,33 В. Таким чином, у першому фоні окреме виявлення йонів нікелю і цинку неможливе, а в другому можливе.
4.2.5. Кількісний полярографічний аналіз.
Кількісний полярографічний аналіз грунтується на залежності граничного дифузійного струму від концентрації визначуваної речовини, яка виведена Ільковичем:
I |
гр |
605nCD 12 m |
23 16 |
, |
(4.22) |
|
|
|
|
|
де Ігр - сила граничного дифузійного струму, мка, С - концентрація визначуваної речовини, ммоль/л, D - коефіцієнт дифузії, см2/c,
m - швидкість витікання ртуті, мг/с,- період капання ртуті, с.
72