

ПРИЛОЖЕНИЕ № 2. ГЕННЫЕ БОЛЕЗНИ
1. Причины ферментопатии. Генные мутации у человека являются причинами многих наследственных моногенных заболеваний. При изучении белковых продуктов мутантных генов выделяют две группы мутаций. Первая группа связана с качественными изменениями белковых молекул, т.е. наличием у больных аномальных белков (например, аномальные гемоглобины), что обусловлено мутациями структурных генов. Другая группа заболеваний характеризуется количественными изменениями содержания нормального белка в клетке (повышение или снижение), что обусловлено, чаще всего, мутациями функциональных генов, т.е. связано с нарушениями регуляции работы генов.
Эти нарушения могут осуществляться на различных уровнях:
претранскрипционном – это осуществляется путем увеличения или уменьшения числа копий гена,
транскрипционном – это генетические дефекты в спейсерах, интронах, транспозонах, регуляторных белках могут приводить к нарушению транскрипции всего гена, обусловливающие изменение объема синтеза соответствующего белка,
процессинга и сплайсинга про-и-РНК - это нарушения на уровне разрушения неинформативных участков про-и-РНК и сплавления информативных участков,
трансляционном – это нарушения на уровне непосредственной сборки белковой молекулы в рибосоме,
посттрансляционном – это нарушения на уровне образования вторичной, третичной и четвертичной структуры белковой молекулы.
Генные мутации фенотипически проявляются как наследственные болезни обмена веществ — ферментопатии. Вещества, накапливающиеся в результате отсутствия или снижения активности ферментов, либо сами оказывают токсическое действие, либо включаются в цепи вторичных обменных процессов, в результате которых образуются токсические продукты. В настоящее время описано около 3 тыс. наследственных болезней обмена веществ. Общая частота генных болезней в популяциях людей составляет 2—4%.
Генные болезни классифицируют по их фенотипическому проявлению, т.е. болезни, связанные с нарушением обменов:
аминокислотного,
углеводного,
липидного,
минерального,
нуклеиновых кислот,
в системе:
свертывания крови,
гемоглобинопатии,
развития соединительной ткани и др.
2. Разберем наиболее часто встречающиеся болезни, связанные с нарушением аминокислотного обмена: фенилкетонурию и альбинизм.
В норме аминокислота фенилаланин (ФА) с помощью фермента фенилаланингидроксилазы превращается в аминокислоту — тирозин, который в свою очередь под действием фермента тирозиназы может превращаться в меланин (пигмент). В результате нарушения активности этих ферментов развиваются два наследственных заболевания обмена человека: фенилкетонурия и альбинизм.
|
ФЕНИЛ- АЛАНИН
ФПВК |
-гидрокси-лаза
|
ТИРОЗИН |
т |
МЕЛАНИН
|
|
МЕЛАНИН |
|
АЛЬБИНИЗМ
|
| |||||
|
ТАНУРИЯ | ||||||
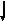
 фенилаланин
фенилаланин ирозиназа
ирозиназа

 ФЕНИЛКЕ-
ФЕНИЛКЕ-Схема нормального обмена и его нарушений аминокислоты фенилаланина
2.1 Фенилкетонурия (ФКУ) встречается в человеческих популяциях с относительно высокой частотой — 1:10000. Заболевание наследуется по аутосомно-рецессивному типу — больные являются рецессивными гомозиготами (аа). Механизм возникновения – мутация в системе генов, отвечающих за синтез фермента фенилаланин-гидроксилазы.
Фенилаланин принадлежит к числу незаменимых аминокислот. Только часть ФА используется для синтеза белков; основное количество этой аминокислоты окисляется до тирозина. Если неактивен фермент фенилаланингидроксилаза, то ФА не превращается в тирозин, а накапливается в сыворотке крови в больших количествах и превращается в фенилпировиноградную кислоту (ФПВК), выделяющуюся с мочой и потом, вследствие чего от больных исходит «мышиный» запах. Дети с фенилкетонурией рождаются здоровыми (фенотипически), но в первые недели жизни у них появляются клинические признаки заболевания. ФПВК является нейротропным ядом, в результате чего развивается повышенная возбудимость, повышенный тонус мышц, судорожные припадки. Позже присоединяются нарушения высшей нервной деятельности, умственная отсталость, микроцефалия. У больных наблюдается слабая пигментация вследствие нарушения синтеза меланина.
Диагностика ФКУ осуществляется биохимическими методами:
еще до развития клинической картины наблюдается выделение ФПВК с мочой (хлорное железо, мазок по пеленки),
в крови определяется высокое содержание фенилаланина.
Метод лечения ФКУ является диетотерапия — снижение содержания в пище ребенка аминокислоты фенилаланина (специальные смеси с отсутствием фенилаланина, а позднее с небольшим добавление этой аминокислоты). Лечение необходимо начинать с первых недель жизни, проводить до 7—10 лет и постоянно следить за содержанием фенилаланина в крови (у человека до 7 лет идет формирование нервной системы). Мозг взрослого человека устойчив к высоким концентрациям ФПВК.
2.2. Альбинизм встречается с частотой — от 1:5000 до 1:25000. Он наследуется по аутосомно-рецессивному типу. Механизм этого заболевания - мутация гена, при которой нарушается активность фермента тирозиназы, превращающего аминокислоту тирозин в пигмент меланин.
Основными клиническими проявлениями альбинизма являются отсутствие меланина в клетках кожи (молочно-белый ее цвет), волосах (очень светлые волосы), радужной оболочке глаза и повышенная чувствительность к УФ облучению, которое вызывает воспалительные заболевания кожи. У больных на коже отсутствуют какие-либо пигментные пятна, снижена острота зрения. Фенотипические признаки выражены уже у новорожденного.
Диагностика заболевания по фенотипическим признакам.
Лечение не разработано (защита от солнечных лучей, поднятие иммунитета).
2.3. Алкаптонурия встречается довольно редко (3-5/106). Наследуется по аутосомно-рецессивному типу. Алкаптонурия является следствием мутации гена регулирующего синтез оксидазы, катализирующей превращение гомогентизиновой кислоты в малеилацетоуксусную.
В основе этой патологии лежат отложения гомогентизиновой кислоты в соединительной ткани.
Диагностика:
клиника (фенотипические проявления: потемнение мочи на воздухе до цвета охры) проявления заболевания начинаются после 40 лет, характеризуются поражением суставов конечностей и позвоночника;
биохимические методы.