

Tools and Applications of Biochemical Engineering Science
.pdfProduction of Core and Virus-like Particles with Baculovirus Infected Insect Cells |
201 |
of the stirring rate. This also implies that when the process is scaled-up to a large scale, keeping the shear stress constant, the overall infection rate should be the same independent of the higher turbulence regime at the larger scale [98].
From the Dee and Shuler model the virus depletion rate function can be defined as [106]:
dVex |
= − ka .C.Vex |
(1) |
|
||
dt |
|
|
Where C is the cell concentration (cells/ml),Vex is concentration of free virus in suspension (pfu/ml) and ka is the attachment constant (ml/cells h–1).
To know the binding kinetics is also important in MOI-TOI specification, in particular at low MOI’s, for different cell lines. T. ni cells like HighFive‘ have a constant of attachment almost 10-fold higher than S. frugiperda cells such as Sf9 [60].
Considering equal time steps (Dt) Dee and Shuler propose that the amount of virus bound during the first time step could be approximated by:
∆ V1 = ka C1V0∆ t |
(2) |
Where C1 is the cell concentration after time step 1.
In the Licari and Bailey Model [102] and also in the latest Hu and Bentley
Model [105] |
it is proposed that the infection process be described by |
the Poisson |
distribution with mean and variance equal to a.MOI. The |
a-value has been proposed to be dependent on the physical system and a value of a = 0.04 was proposed for static systems [102]. For agitated systems suspension cultures Hu and Bentley proposed a value of a = 0.08 because they state that agitation systems enable higher efficiency of contact between viruses and cells [105]. This is not absolutely true, at least the true reason is not the higher mixing level but the fact that in static cultures, less cell surface is exposed to the virus, since to the cells are attached to a surface. This gives an overall constant of attachment 3-4 fold lower than in suspension systems [61].
Licari and Bailey used the Poisson distribution in the following form:
p(t, j) = |
e− α .MOI( t) × |
(α .M O I(t)) j |
(3) |
|
j! |
||
|
|
|
p(t, j) is the probability at time t that a cell could be infected by j viruses, t is the time (h), j is the number of viruses infecting one cell, a is the empirical factor and MOI(t) is the Multiplicity of Infection at time t.
The sum of all possible combinations of the number of viruses that could be infecting one cell is given by:
P(t) = ∑∞ |
p(t, j) = 1− e− α .MOI( t) |
(4) |
j= 1 |
|
|
202 |
M.J.T. Carrondo et al. |
Dee and Shuler [106] applied the same Poisson distribution but with a mean and variance equal to:
λ = |
∆ V1 |
= |
ka C1V0∆ t |
= k |
C |
M O I ∆ t |
(5) |
|
|
||||||
|
C1 |
|
a |
0 |
0 |
|
|
|
C1 |
|
|
|
|||
Where MOI0 represents the multiplicity of infection at time 0. Comparing the Licari and Bailey parameter a with the Dee and Shuler expression means that:
α = ka C0∆ t |
(6) |
If we take the value of ka = 0.44¥10–9 (ml/cells min) proposed by Wickham et al. [60] for static cultures with C0 = 1.5¥106 cells/ml (mid-exponential curve of Licari and Bailey data [102]) and a time step Dt = 1 h = 60 min, the computed value of a is 0.04, in agreement with Licari and Bailey [102].
Also if we apply the same concept, to the data available for suspension cultures, with ka = 1.3¥10–9 (ml/cells min), proposed by Dee and Shuler [61], with C0 = 1.0¥106 cells/ml (early-exponential curve of Hu and Bentley data [33]) and a time step Dt = 1 h = 60 min, the computed value of a is 0.08.
Nevertheless, the parameter with real physical meaning is ka and not a, since ka is a real constant that has been measured, contrary to the case of the empirical parameter a. Licari and Bailey have used an assumption that the a value will remain constant for all initial infections since the physical system and the volume used are the same [102]. This is likely not to be the case since as can be seen by the expression above, the a value is dependent upon cell concentration. Also in the Hu and Bentley Model they propose that a will remain constant over time [105].
Secondary infection is an issue that is dealt with only in the Dee and Shuler Model [61] and Hu and Bentley Model [105]. Since today there is not sufficient knowledge regarding the number of viruses that are really infecting a single cell, the linear approach taken by Hu and Bentley [105] for cell “infectivity” status is an approach as good as any other. However, Hu and Bentley assume that all viruses initially infecting cells are effective (not degraded), while all viruses that secondarily infect the already-infected cells are defective and degraded after entering the cells; these authors claim that approximates the formulation of Dee and Shuler [61] in a mathematically tractable form. Although mathematically this appears equivalent it is not obvious that this assumption correlates well with the effect of the number of viruses infecting a single cell has on its specific productivity. As suggested by Power and Nielsen [107] since these measurements can not be validated, the model that arises with this kind of formalism becomes a sophisticated fitting machine.
5.2
Production of VLPs
All models developed dealt with product expression by a sub-division of cellular events and a more or less linear relationship between product expression rates and infected cell concentrations.Although this is a simplification, since the
Production of Core and Virus-like Particles with Baculovirus Infected Insect Cells |
203 |
expression occurs in a transient mode, the models in general fit the data with good correlation.
The Hu and Bentley model is the only one that tries to describe VLP production and assembling in baculovirus infected insect cells [105]. Nevertheless, regarding VLP assembly, the formalism presented is completely theoretical and based on the assembly pathway of icosahedral viruses. From a process development point of view, this model does not generate enough output to make it applicable to bioreaction operational parameters definition. However, it can be used as the basis for a more structured approach to the VLP assembling process in baculovirus infected insect cells.
The application of mathematical modelling to baculovirus infection and vi- rus-like particle production was also successfully done to Parvovirus B19 viruslike particle production with two different baculovirus at low MOI’s [18]. But in this model the same concepts proposed in the Licari and Bailey Model was applied, i.e. baculovirus infection follows Poisson distribution with mean and variance equal to a.MOI, but with co-infection with two single-vectors, each one encoding a specific viral protein.
However, as important as the Hu and Bentley Model is the stepwise approach to process optimisation that Hu and Bentley have reported [33]. The focus on quantitative analysis of protease degradation of the product over time, along with the similar approach followed by Cruz et al. [25], also indicate new directions to follow in mathematical modelling regarding product expression optimisation.
Mathematically modelling of VLP production by baculovirus infected insect cells is not a closed chapter of research, and probably never will be, at least until complete structured models have been developed, taking into account intracellular pools.
James Bailey has said, talking about the inextricable coupling between a model and its intended application (citing Casti) that “Basically the point of making models is to be able to bring a measure of order to our experience and observations, as well as to make specific predictions about certain aspects of the world we experience” [108]. So models are not an end in themselves but should be looked more as tools to think and calculate logically about what components and interactions are important in a complex system. Mathematical models can also be justifiable to minimize the experimental effort, predict system behaviour and identify new research avenues [10].
6 Conclusion
The production of VLPs with baculovirus infected insect cells is entering the phase of industrial application. Due to the complexity of the system a multitude of vector strategies have been developed, ranging from single to fifth-level baculovirus vectors, along with combinations between them.
Bioreaction optimisation involves assessment of good oxygen transfer conditions and evaluation of shear stress influence on cell growth and productivity.
204 |
M.J.T. Carrondo et al. |
Scalability of the systems is a must and the use of low MOI strategies is obligatory if the goal is the industrial application. Complex feeding strategies must be avoided since they impose high risks, and costs, to the overall process.
Better mathematical models providing a better understanding of the basic events are still needed, incorporating the attachment steps, the intracellular events, and product expression, assembling and degradation.
Acknowledgement. The authors acknowledge and appreciate the financial support received from the European Commission (BIO4-CT98-0215) and from Fundação para a Ciência e Tecnologia – Portugal (PRAXIS XXI/BD/16136/98).
References
1.Wu SC, Jarvis DL, Dale BE, Liao JC (1994) Biotechnol Prog 10:55
2.Morais VA, Serpa J, Palma AS, Costa T, Maranga L, Costa J (2001) Biochem J 353:719
3.Casal JI (1996) Cytotechnology 20:261
4.Roy P (1990) FEMS Microbiol Immunol 2:223
5.Cruz PE, Peixoto CC, Moreira JL, Carrondo MJT (1998) J Chem Technol Biotechnol 72: 149
6.Blissard GW (1996) Cytotechnology 20:73
7.O’Reilly DR, Miller LK (1989) Baculovirus expression vectors, a laboratory manual. WH Freeman and Co., New York
8.King LA, Possee RD (1992) The Baculovirus Expression System: a laboratory guide. Chapman Hall, London
9.Touze A, Coursaget P (1998) Nucleic Acids Res 26:1317
10.Cruz PE (1999) PhD Thesis – Optimization of the Production of Complex Bioproducts, PhD Thesis, ITQB-UNL
11.Baumert TF, Ito S, Wong DT, Liang TJ (1998) J Virol 72:3827
12.Hyatt AD, Zhao Y, Roy P (1993) Virology 193:592
13.French TJ, Roy P (1990) J Virol 64:1530
14.French TJ, Marshall JJ, Roy P (1990) J Virol 64:5695
15.Sabara M, Parker M, Aha P, Cosco C, Gibbons E, Parsons S, Babiuk LA (1991) J Virol 65: 6994
16.Roy P (1992) Vet Microbiol 33:155
17.Zheng YZ, Greenfield PF, Reid S (1999) Biotechnol Bioeng 65:600
18.Tsao EI, Mason MR, Cacciuttolo MA, Bowen SH, Folena-Wasserman G (1996) Biotechnol Bioeng 49:130
19.Brown CS, Van Lent JW, Vlak JM, Spaan WJ (1991) J Virol 65:2702
20.Martinez C, Dalsgaard K, Lopez de Turiso JA, Cortes E,Vela C, Casal JI (1992) Vaccine 10: 684
21.Hernando E, Llamas-Saiz AL, Foces-Foces C, McKenna R, Portman IAgbandje-McKenna M, Almendral JM (2000) Virology 267:299
22.Langeveld JP, Kamstrup S, Uttenthal A, Strandbygaard B, Vela C, Dalsgaard K, Beekman NJ, Meloen RH, Casal JI (1995) Vaccine 13:1033
23.Bansal GP, Hatfield JA, Dunn FE, Kramer AA, Brady F, Riggin CH, Collett MS,Yoshimoto K, Kajigaya S, Young NS (1993) Journal of Infectious Diseases 167:1034
24.Jiang B, Barniak V, Smith RP, Sharma R, Corsaro B, Hu B, Madore HP (1998) Biotechnol Bioeng 60:369
25.Cruz PE, Martins PC, Alves PM, Peixoto CC, Santos H, Moreira JL, Carrondo MJ (1999) Biotechnol Bioeng 65:133
26.Naggie S, Bentley WE (1998) Biotechnol Prog 14:227
27.Fan Z, Yang QR, Twu JS, Sherker AH (1999) J Med Virol 59: 131
Production of Core and Virus-like Particles with Baculovirus Infected Insect Cells |
205 |
28.Heile JM, Fong YL, Rosa D, Berger K, Saletti G, Campagnoli S, Bensi G, Capo S, Coates S, Crawford K, Dong C, Wininger M, Baker G, Cousens L, Chien D, Ng P, Archangel P, Grandi G, Houghton M, Abrignani S (2000) J Virol 74:6885
29.Roy P, Bishop DH, LeBlois H, Erasmus BJ (1994) Vaccine 12:805
30.Belyaev AS, Roy P (1993) Nucleic Acids Res 21:1219
31.Belyaev AS, Hails RS, Roy P (1995) Gene 156:229
32.Cruz PE, Peixoto CC, Devos K, Moreira JL, Saman E, Carrondo MJT (2000) Enzyme Microb Technol 26:61
33.Hu YC, Bentley WE (1999) Biotechnol Prog 15:1065
34.Loudon PT, Hirasawa T, Oldfield S, Murphy M, Roy P (1991) Virology 182:793
35.Le Blois H, Fayard B, Urakawa T, Roy P (1991) J Virol 65:4821
36.Belyaev AS, Roy P (1992) Virology 190:840
37.Sedlik C, Sarraseca J, Rueda P, Leclerc C, Casal I (1995) J Gen Virol 76 ( Pt 9):2361
38.Sedlik C, Saron M, Sarraseca J, Casal I, Leclerc C (1997) Proc Natl Acad Sci USA 94: 7503
39.Rueda P, Martinez-Torrecuadrada JL, Sarraseca J, Sedlik C, del Barrio M, Hurtado A, Leclerc C, Casal JI (1999) Vaccine 18:325
40.Hu YC, Bentley WE, Edwards GH, Vakharia VN (1999) Biotechnol Bioeng 63:721
41.Wang MY, Kuo YY, Lee MS, Doong SR, Ho JY, Lee LH (2000) Biotechnol Bioeng 67:104
42.Bhatia R, Jesionowski G, Ferrance J, Ataai MM (1996) Cytotechnology 20:33
43.Schmid G (1996) Cytotechnology 20:43
44.Schlaeger E-J (1996) Cytotechnology 20:57
45.Grace TDC (1962) Nature 195:788
46.Hink WF (1976) Invertebrate Tissue Culture Research Applications. Academic Press, New York p 319
47.Hink WF, Hall RL (1989) In: Mitsuhashi J (ed) Invertebrate Cell System Applications, vol II. CRC Press, Inc., Boca Raton, FL, p 269
48.Baines D (1996) Cytotechnology 20:13
49.Agathos SN, Jeong YH, Venkat K (1990) Ann N Y Acad Sci 589:372
50.Parker SD, Hunter E (2000) J Virol 74:784
51.Vaughn JL, Goodwin RH, Tompkins GJ, McCawley P (1977) In Vitro 13:213
52.Cruz PE, Moreira JL, M.J.T. C (1997) Biotechnology Techniques 11:117
53.Wickham TJ, Davis T, Granados RR, Shuler ML, Wood HA (1992) Biotechnol Prog 8:391
54.Wickham TJ, Nemerow GR (1993) Biotechnol Prog 9:25
55.Davis TR, Wickham TJ, McKenna KA, Granados RR, Shuler ML, Wood HA (1993) In Vitro Cell Dev Biol Anim 29A:388
56.Agathos SN (1996) Cytotechnology 20:173
57.Gheysen D, Jacobs E, de Foresta F, Thiriart C, Francotte M, Thines D, De Wilde M (1989) Cell 59:103
58.Aunins JG (2000) In: Spier RE, Editor in Chief, The Encyclopedia of Cell Technology. Wiley, New York, p 1182
59.Wickham TJ, Granados RR,Wood HA, Hammer DA, Shuler ML (1990) Biophys J 58:1501
60.Wickham TJ, Shuler ML, Hammer DA, Granados RR, Wood HA (1992) J Gen Virol 73: 3185
61.Dee KU, Shuler ML (1997) Biotechnol Bioeng 54:468
62.Bedard C, Tom R, Kamen A (1993) Biotechnol Prog 9:615
63.Ohman L, Ljunggren J, Haggstrom L (1995) Appl Microbiol Biotechnol 43:1006
64.Ferrance JP, Goel A, Ataai MM (1993) Biotechnol Bioeng 42:697
65.Wong TK, Nielsen LK, Greenfield PF, Reid S (1994) Cytotechnology 15:157
66.Hensler WT, Agathos SN (1994) Cytotechnology 15:177
67.Rhiel M, Mitchell-Logean CM, Murhammer DW (1997) Biotechnol Bioeng 55:909
68.Bedard C, Kamen A, Tom R, Massie B (1994) Cytotechnology 15:129
69.Cruz PE, Cunha A, Peixoto CC, Clemente J, Moreira JL, Carrondo MJ (1998) Biotechnol Bioeng 60:408
70.Kloppinger M, Fertig G, Fraune E, Miltenburger HG (1990) Cytotechnology 4:271
206 |
M.J.T. Carrondo et al. |
71.Caron AW, Tom RL, Kamen AA, Massie B (1994) Biotechnol Bioeng 43:881
72.Deutschmann SM, Jager V (1994) Enzyme Microb Technol 16:506
73.Fertig G, Rahn HP, Angermann A, Kloppinger M, Miltenburger HG (1993) Cytotechnology 11:67
74.Jäger V (1996) Cytotechnology 20:191
75.Chan LC, Greenfield PF, Reid S (1998) Biotechnol Bioeng 59:178
76.Nguyen B, Jarnagin K, Williams S, Chan H, Barnett J (1993) Journal Biotechnol 31:205
77.van Lier FL, van den End EJ, de Gooijer CD, Vlak JM, Tramper J (1990) Appl Microbiol Biotechnol 33:43
78.Piergiovani PR, Dutt K (1993) Biotechnology Techniques 7:765
79.Wang MY, Bentley WE (1994) Appl Microbiol Biotechnol 41:317
80.Maiorella B, Inlow D, Shauger A, Harano D (1988) Bio/Technology 6:1406
81.Reuveny S, Kim YJ, Kemp CW, Shiloach J (1993) Appl Microbiol Biotechnol 38:619
82.Hong-Liang S, Zuo-Hu L (1998) Appl Environ Microbiol 64:2237
83.Pham MQ, Naggie S, Wier M, Cha HJ, Bentley WE (1999) Biotechnol Bioeng 62:175
84.Donaldson M, Wood HA, Kulakosky PC, Shuler ML (1999) Biotechnol Bioeng 63:255
85.Medina M, Domingo E, López-Rivas A, Zuidema D, Vlak JM, Belsham GJ (1995) Cytotechnology 17:21
86.Elias CB, Zeiser A, Bedard C, Kamen AA (2000) Biotechnol Bioeng 68:381
87.Scott RI, Blanchard JH, Ferguson CH (1992) Enzyme Microb Technol 14:798
88.Konz JO, King J, Cooney CL (1998) Biotechnol Prog 14:393
89.Wang MY, Kwong S, Bentley WE (1993) Biotechnol Prog 9:355
90.Palomares LA, Ramirez OT (1996) Cytotechnology 22:225
91.Taticek RA, Shuler ML (1997) Biotechnol Bioeng 54:142
92.Michaels JD, Mallik AK, Papoutsakis ET (1996) Biotechnol Bioeng 51:399
93.Murhammer DW, Goochee CF (1988) Bio/Technology 6:1411
94.Murhammer DW, Goochee CF (1990) Biotechnol Prog 6:391
95.Chalmers JJ (1996) Cytotechnology 20:163
96.Tatterson GB (1991) Fluid mixing and gas dispersion in agitated tanks. MacGraw-Hill, Inc., New York
97.Tramper J, Vlak JM, Gooijer CDd (1996) Cytotechnology 20:221
98.Maranga L, Cunha A, Clemente J, Carrondo MJT (2000) Conference Proceedings of Biotechnology 2000 – The World Congress in Biotechnology. Dechema, Berlin, p 392
99.Krell PJ (1996) Cytotechnology 20:125
100.Wong KTK, Peter CH, Greenfield PF, Reid S, Nielsen LK (1996) Biotechnol Bioeng 49:659
101.Radford KM, Cavegn C, Bertrand M, Bernard AR, Reid S, Greenfield PF (1997) Cytotechnology 24:73
102.Licari P, Bailey JE (1992) Biotechnol Bioeng 39:432
103.Wu S, Dale BS, Liao JC (1993) Biotechnol Bioeng 41:104
104.Power J, Greenfield PF, Nielsen L, Reid S (1992) Cytotechnology 9:149
105.Hu Y-C, Bentley WE (2000) Chem Eng Sci 55:3991
106.Dee KU, Shuler ML (1997) Biotechnol Prog 13:14
107.Power JF, Nielsen LK (1996) Cytotechnology 20: 209
108.Bailey JE (1998) Biotechnol Prog 14:8
109.Lanford RE, Luckow V, Kennedy RC, Dreesman GR, Notvall L, Summers MD (1989) J Virol 63:1549
110.White LJ, Hardy ME, Estes MK (1997) J Virol 71:8066
111.Kirnbauer R, Booy F, Cheng N, Lowy DR, Schiller JT (1992) Proc Natl Acad Sci USA 89: 12180
112.Montross L,Watkins S, Moreland RB, Mamon H, Caspar DL, Garcea RL (1991) J Virol 65: 4991
113.Kosukegawa A, Arisaka F, Takayama M, Yajima H, Kaidow A, Handa H (1996) Biochim Biophys Acta 1290:37
Received: May 2001

Integrated Approach To Explore the Potential of Marine Microorganisms for the Production of Bioactive Metabolites
Irene Wagner-Döbler1, Winfried Beil2, Siegmund Lang3,*, Marinus Meiners4,
Hartmut Laatsch5
1Gesellschaft für Biotechnologische Forschung, Mascheroder Weg 1, 38124 Braunschweig, Germany
E-mail: iwd@gbf.de
2Institut für Allgemeine Pharmakologie, Medizinische Hochschule Hannover, Konstanty-Gutschow-Strasse 8, 30625 Hannover, Germany
E-mail: beil.winfried@mh.hannover.de
3Institut für Biochemie und Biotechnologie, TU Braunschweig, Spielmannstrasse 7, 38106 Braunschweig, Germany
E-mail: s.lang@tu-bs.de
4Fachbereich Naturwissenschaftliche Technik, Fachhochschule Ostfriesland, Constantiaplatz 4, 26726 Emden, Germany
E-mail: meiners@nt-newton.fho-emden.de
5Institut für Organische Chemie, Universität Göttingen, Tammannstrasse 2, 37077 Göttingen, Germany
E-mail: hlaatsc@gwdg.de
Dedicated to Prof. Dr. Wolf-Dieter Deckwer on the occasion of his 60th birthday
During the last 10 years marine organisms have provided a large number of new natural products. Interesting compounds have mainly been derived from macroorganisms such as sponges, ascidians, corals and bryozoans. The number of secondary metabolites from marine microorganisms is smaller, but rapidly increasing. Because of the enormous difficulties involved in harvesting products from marine animals, and the fact that some of the bioactive compounds are produced by associated bacteria, the advantages of sustainable production of bioactive metabolites by bacteria or fungi, under the protection of natural resources, seem to be very attractive for the future. This review describes current progress in the isolation and identification of novel marine microorganisms, the discovery of new secondary metabolites, the biotechnological approaches to overproduce them, as well as the evaluation and characterization of their bioactivity.
Keywords. New bacteria from the North Sea, Secondary metabolites, Bioactivity, Biotechnological production, Structure elucidation, Phylogenetic screening
1Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 208
1.1Marine Bacteria as Sources of New Bioactive Compounds . . . . . . 208
1.2 |
The Phylogenetic Tree and the Production of Bioactive Compounds |
210 |
1.3 |
Research Network Marine Biotechnology in Lower Saxony . . . . . |
211 |
* To whom all correspondence should be addressed.
Advances in Biochemical Engineering/
Biotechnology, Vol. 74
Managing Editor: Th. Scheper
© Springer-Verlag Berlin Heidelberg 2002
208 |
S. Lang et al. |
2Molecular Tools for Phylogenetic Screening of the Marine Microbial Diversity and First Results on an Indigenous Group of Marine
|
Microorganisms . . . . . . . . . . . . . . . . . . . . . . . . |
. . . . . 212 |
2.1 |
Isolation of Marine Bacteria . . . . . . . . . . . . . . . . . . |
. . . . . 212 |
2.2 |
Phylogenetic Screening of Marine Bacteria . . . . . . . . . |
. . . . . 213 |
2.3 |
Screening of Genetic Diversity – Genomic Fingerprints . . |
. . . . . 214 |
2.4 |
The Marine Roseobacter Clade of the Proteobacteria . . . . |
. . . . . 215 |
3 |
Screening for Novel Products and New Capabilities . . . . |
. . . . . 218 |
4 |
Strategies for Identifying Anticancer Compounds . . . . . |
. . . . . 219 |
4.1 |
Anticancer Drug Development . . . . . . . . . . . . . . . . |
. . . . . 220 |
4.1.1 |
In Vitro Tests . . . . . . . . . . . . . . . . . . . . . . . . . . |
. . . . . 220 |
4.1.2 |
In Vivo Antitumor Tests . . . . . . . . . . . . . . . . . . . . |
. . . . . 221 |
4.1.3 |
Preclinical Pharmacokinetics . . . . . . . . . . . . . . . . . |
. . . . . 221 |
4.1.4 |
Preclinical Toxicology . . . . . . . . . . . . . . . . . . . . . |
. . . . . 222 |
4.2 |
The Screening of Extracts from North Sea Microorganisms |
. . . . . 222 |
5Biotechnological Studies To Overproduce Metabolites
|
by Marine Microorganisms . . . . . . . . . . . . . . . . . . . . . . . |
224 |
5.1 |
Overview of Biochemical Engineering Approaches . . . . . . . . . . |
224 |
5.2 |
Cultivation of North Sea Bacteria . . . . . . . . . . . . . . . . . . . . |
226 |
6 |
Chemical Structures . . . . . . . . . . . . . . . . . . . . . . . . . . . |
228 |
6.1 |
Dereplication . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
228 |
6.2 |
Structure Elucidation and Results . . . . . . . . . . . . . . . . . . . |
229 |
7 |
Concluding Remarks . . . . . . . . . . . . . . . . . . . . . . . . . . |
234 |
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . |
235 |
|
1 Introduction
1.1
Marine Bacteria as Sources of New Bioactive Compounds
The search for bioactive compounds is presently reaching a new dimension – with such diverse approaches as genomics, proteomics, bioinformatics, combinatorial biosynthesis, combinatorial chemistry, targeted drug development, directed evolution of key enzymes, phage display libraries, automation and high throughput screening [1–4]. What role can traditional natural product screening play in today’s drug development approaches [5], and what in particular
Integrated Approach to Explore the Potential of Marine Microorganisms |
209 |
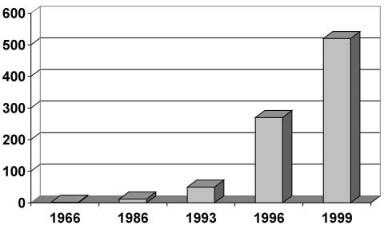
Fig. 1. Total number of metabolites from marine microorganisms (1966–1999)
can be expected from marine microorganisms [6, 7] as sources of novel compounds?
Marine organisms have provided a seemingly endless parade of novel structures. New carbon skeletons were described with a frequency that exceeded all expectations. Several structural features are uniquely or predominantly marine (reviewed by Faulkner since 1986 [8–10]). Thus, ample raw material was provided for combinatorial chemistry, synthetic chemistry, and biocatalysis [11]. The most striking change in the direction of marine natural product chemistry since 1993 was reflected in the sudden increase in reports of new metabolites from marine microorganisms (Fig. 1): Whereas less than ten compounds had been known 20 years ago [12], the number of structures has exponentially grown since then. Although a total number of around 400 compounds is still small in comparison with more than 15,000 structures from terrestrial microorganisms, new compounds are presently almost exclusively found in marine organisms (W Fenical, personal communication).
A number of invertebrate phyla is restricted to the marine environment, with no or few representatives in aquatic and terrestrial habitats, e.g., the Ascidia, Porifera, Coelenterata and Bryozoa. Consequently, from these organisms, a whole range of new chemical structures has been discovered that are not found in terrestrial organisms. However, in many cases, it is not yet clear whether the bioactive compound is produced by the invertebrate, by endosymbiontic or epiobiotic bacteria, or in a cooperative way by both. There is increasing evidence for an important role of the bacterial endosymbionts in the bryostatinproducing bryozoon Bugula neritina [13, 14] or for metabolite production in demospongiae [15].
210 |
S. Lang et al. |
1.2
The Phylogenetic Tree and the Production of Bioactive Compounds
What are marine bacteria? Defining them as bacteria with an absolute requirement for sodium chloride is not a practical solution, because many marine isolates may tolerate quite a wide range of salinities, prompting speculation that they are in fact terrestrial organisms that have been swamped into the oceans from rivers, estuaries and sewage outfalls. Pragmatically, marine microorganisms are therefore defined as bacteria that have been isolated from marine sources on marine media [16].
However, one has to bear in mind that the microbial ecology of marine habitats has been revolutionized by cultivation-independent analyses based on 16S rRNA. It is now well documented that only a fraction of the marine microbial diversity has been cultivated, presumably far less than 1% [17], no more than the “tip of the iceberg” [18]. Clone libraries of marine bacterioplankton 16S rRNA genes are dominated by a few phylotypes that have not been cultivated to date, and which are distributed globally [19, 20]. It can therefore be concluded that the “true” marine microorganisms are in most cases presently not known.
Within the operationally defined “marine bacteria”, i.e., bacteria isolated from marine samples on marine media, bioactive compounds have been reported from Pseudoalteromonas, Cytophaga, Alteromonas, Micrococcus, Bacillus, Acinetobacter, Agrobacterium and Pseudomonas or from unidentified bacteria (Fig. 2).
Cultivated marine bacteria are scattered throughout the phylogenetic tree of the domain Bacteria. However, at lower phylogenetic levels, clusters of marine bacteria have been found which are distinct from those of terrestrial origin. One example is the so-called a3-subgroup of the a-Proteobacteria subclass of the division Proteobacteria, the Roseobacter clade [20]. A marine group of Actinobacteria [21] has been described, which has, to date, however not been cultivated.
Presently, there are three microbial phylogenetic hot spots known for the production of secondary metabolites:
1.The Streptomycetes, a group of filamentous Gram-positive bacteria (Actinomycetes) that are the work horses of natural product isolation [22];
2.the Myxobacteria, motile bacteria with a complex life cycle which form a distinct cluster within the d-subclass of the Proteobacteria and have shown to be a rich source of novel structures and biological activities [23, 24];
3.the Cyanobacteria, the former bluegreen algae, photosynthetic bacteria which are distributed globally and produce extremely potent toxins (e.g., [25–27]).
In addition, the antibiotics and other bacteriocins were originally detected in lactic acid bacteria, but were later also found in other Gram-positive microorganisms [28]. Lactic acid bacteria are a group of non-spore-forming, anaerobic fermentative bacteria within the Gram-positives with low GC content.