

New Products and New Areas of Bioprocess Engineering
.pdfMultistage Magnetic and Electrophoretic Extraction of Cells, Particles and Macromolecules |
171 |
2.Systems in which an organic reverse micellar solution is in equilibrium with a conjugated aqueous phase.
Both the systems are based on the differential partitioning of biomolecules between the immiscible phases. However, the principles of solubilization differ greatly. These two developing techniques will be discussed briefly here.
3.1
Existing Methods – Brief Analysis
Even though aqueous two-phase extraction (ATPE) has been known for quite some time, it has gained importance for industrial exploitation only recently. Unlike conventional liquid-liquid extraction involving organic and aqueous phases or pairs of organic phases, ATPE employs two aqueous phases. ATPE has been successful to a large extent in overcoming the limitations of conventional extraction such as poor solubility of proteins in organic solvents and the tendency of organic solvents to denature proteins/enzymes. ATPE has been recognized as a versatile technique for downstream processing of biomolecules such as proteins, enzymes, viruses, cells, cell organelles, and other biological materials [70, 80–82]. Its applications for cells and particles was discussed in the earlier sections. ATPE offers many advantages such as biocompatible environment, low interfacial tension, low energy, easy scale-up, and continuous operation. More importantly, partitioning depends on differences in surface properties and does not depend on size, shape, or density of the separand except under specific conditions [78, 83]. Further, the equipment and methods of conventional organic aqueous phase extraction used in the chemical industry can be easily adapted to ATPE. However,ATPE is not selective enough to provide the extreme purity usually desired. The main reasons for ATPE not reaching industry are perhaps the high cost of the phase forming polymers and slow demixing rate of the phases. Thus, ATPE has been recognized as a potential primary purification step in the overall protein/enzyme recovery train [84, 85] in which the final purification is achieved by methods such as chromatography or crystallization. ATPE is also effective and efficient for the removal of contaminating materials and undesirable byproducts such as nucleic acids and polysaccharides [83].
Temperature induced phase separation, proposed by Galaev and Mattiasson [86] and studied extensively by Tjerneld and coworkers [87–90] enables the recovery and recycling of the polymers so the economics have improved to a large extent. It also improves the yield, degree of selectivity, etc. Methods for faster demixing of the phases were developed by employing electric fields [91, 92] and by acoustic fields [93]. In the near future ATPE is expected to enjoy further commercial adaptation.
Some recent developments in this area, which resulted in either increase in selectivity or improvement in yield, are briefly discussed here. Affinity partitioning (AP) is based on the preferential/biospecific interaction between the molecule and affinity polymer derivative which results in a biomoleculepolymer derivative complex which selectively partitions to one of the phases leaving the contaminating substances or proteins in the other phase. Most of the reported investigations regarding affinity partitioning pertain to polymer/

172 K.S.M.S. Raghavarao et al.
polymer type aqueous two-phase systems (ATPSs) [78] and very few reports are available on polymer/salt type ATPSs, [94] mainly due to the interference of high salt concentrations with the biospecific interactions. More details such as the theory available for AP, future trends, and its other applications are given elsewhere [84, 95, 96].
Metal affinity partitioning exploits the affinity of transition metal ions for electron-rich amino acid residues, such as histidine and cysteine, accessible on the surfaces of proteins. When the metal ion is partially chelated and coupled to a linear polymer, such as polyethylene glycol (PEG), the resulting polymerbound metal chelate can be used to enhance the partitioning of metal binding proteins into the polymer-rich phase of a PEG-salt or PEG-dextran (DX) ATPS. Since most proteins favor the salt-rich heavy phase of an aqueous two-phase system, metal affinity partitioning can be a very efficient and selective means of isolating and purifying a metal-binding protein from a crude mixture [97].
In most cases, the desired protein has been isolated in a single chromatographic step from clarified cell lysate, without further pretreatment [98]. Guinn [95] and Sulkowski [99] successfully achieved the partitioning of recombinant hemoglobin from crude cell lysate using Cu (II) IDA-PEG in a two-phase system of PEG and magnesium sulfate. To our knowledge, this is the first recorded successful attempt to apply metal affinity partitioning techniques to the isolation of a recombinant protein from crude cell lysate. Successful demonstration of this technology sets the stage for its potential commercial use in the isolation of native and non-native metal-binding proteins. More details in this area can be found in a recent review article [96].
Extractive fermentation/bioconversion involves the integration of fermentation/bioconversion with one or more downstream processing step(s), such as extraction using ATPS, ultrafiltration, etc.An opportunity is thus created to explore new types of industrially relevant bioreactor designs [100, 101]. Many examples are given in the review articles by Diamond and Hsu [78] and Raghavarao et al. [79]. Most of the bioconversions have been performed using polymer/polymer type ATPSs. Lee and Chang [102] were among the first to employ the PEG/ potassium phosphate system successfully for the production of acrylamide from acrylonitrile using Brevibactrium sp. The selective partitioning of the product into the top phase was found to reduce the inhibition of the active bacterial enzyme by both the substrate and the product. The same strategy is gaining popularity under the name of aqueous two-phase fermentation (ATPF) [103, 104].
Over the past few years there has been considerable interest in the use of the micro-gravity environment of earth orbit as a laboratory for understanding the role of surface forces in liquid-liquid phase separation. The importance of surface wetting forces on the phase separation of ATPSs in microgravity was observed during Space Shuttle flight STS-26 in October 1988. Separation experiments were performed in a Plexiglas hand-held phase partition experiment (PPE) module consisting of 18 chambers filled with PEG and DX in plastic extraction cavities [105, 106]. Evidence of surface tension-driven phase separation in microgravity and its potential processing advantages led to the development of a reusable platform, the ORSEP, for conducting multi-stage extraction using ATPSs for both terrestrial and space-based processing applications [95, 96].
Multistage Magnetic and Electrophoretic Extraction of Cells, Particles and Macromolecules |
173 |
In microgravity, the coalescence of dispersed phase droplets results in a decrease in dispersed phase volume. Since the continuous phase is incompressible, the decrease in droplet volume induces a flow of the continuous phase toward the droplet. In the absence of buoyancy forces, assuming the dispersed phase droplets are uniformly distributed, there would be an increase in the collision efficiency of droplets due to the effect of local flow. Thus in microgravity, as in unit gravity, coalescence and phase separation is spontaneous and enhanced by the local flow induced by droplet coalescence. This would help to explain why phase separation was possible for some of the systems in STS-26 using the PPE module but not for others and why low viscosity and surface tension had such a large positive effect on demixing rates [107]. Due to the combined effects of Brownian motion and the local flow induced by droplet coalescence there is a greater probability that PEG-rich phase droplets (typically the dispersed phase in PEG/DX systems) would impact the container walls and remain there. If the continuous phase were DX it would prevent the dispersed phase from contacting the container walls. The sole effect of surface wetting forces in microgravity would seem to be giving the phase separation directionality.
Reverse micellar extraction (RME) has been gaining popularity as an attractive liquid-liquid extraction process [108–111]. This is mainly due to the fact that enzymes can be solubilized in organic solvents with the aid of reverse micellar aggregates [112, 113]. Their inner core contains an aqueous microphase, which is able to solubilize polar substances, e.g., hydrophilic enzymes [114]. In many cases not only the enzymes retained their activity in organic environment; in some cases they seem to perform even better if they are entrapped into reverse micellar aggregates [111]. One of the remarkable findings that gave this field a major boost is that the solubilization of different proteins into micellar solutions is a selective process [112].
3.2
Magnetic and Electro-Extraction Methods
3.2.1
Magnetic Extraction
Affinity techniques appear currently to be among the most powerful tools available for downstream processing both in terms of their selectivity and recovery. Conventional porous affinity supports are mostly applicable for work in clarified solutions and not suitable for work in early stages where suspended solids and other fouling compounds are present in the system. Non-porous support particles, which are easier to clean and less prone to fouling, are more useful for the purification from feed streams [29, 115]. However, in order to obtain similar surface area of typical microporous particles of 100 mm, the size of non-porous support particles have to be in the range of 0.1–1 mm [116]. It appears that the only feasible method for the recovery of such small particles in the presence of biological debris of similar size is magnetic separation, which eliminates many time-consuming steps and is easy to carry out. The direct and
174 |
K.S.M.S. Raghavarao et al. |
|
Table 3. Magnetically assisted separation of enzymes |
|
|
|
|
|
Enzyme |
Magnetic system |
Reference |
|
|
|
ADH, LDH, 6-PGDH |
Ferrofluid-modified 5¢-AMP-sephadex 4B |
[117] |
Hexokinase, G-6-PDH |
Magnetic aqueous two-phase system ADH, PTK |
[118] |
Asparaginase |
Magnetic polyacrylamide gel with immobilized |
[119] |
|
d-asparagine |
|
b-Galactosidase |
Silanized magnetite with immobilized |
[119] |
|
p-aminophenyl-b-d-thiogalactopyranoside |
|
Chymostrypsin |
Chitosan-magnetic beads |
[120] |
G-6-PDH |
Ferrofluid-modified 2¢,5¢-ADP-Sepharose 4B |
[121] |
LDH |
Magnetic agarose with immobilized triazine dye |
[122] |
Lysozyme |
Magnetic chitin |
[123] |
Pectinase |
Alginate-magnetite beads |
[124] |
Trypsin |
Magnetic polyurethane-polystyrene beads with |
[125] |
|
immobilized soy trypsin inhibitor |
|
Trypsin |
Sub-micron ferrite particles with immobilized |
[29] |
|
soybean trypsin inhibitor |
|
|
|
|
indirect methods described in the section on cell separation (Sect. 2.1.1) are equally valid for macromolecules as well. Although both the direct and indirect methods perform well, the direct method is generally more controllable. However, the indirect method may perform better for antibodies with poor affinity or when antigen is rare or less accessible [33].
Many magnetic adsorbents have been used for the isolation of various biologically active macromolecules such as enzymes, enzyme inhibitors, DNA, RNA, antibodies, antigens, etc. from different sources such as nutrient media, fermentation broth, tissue extracts, body fluids, and others. However, the present discussion is restricted to enzymes only.
In the case of enzymes, an appropriate affinity ligand is usually immobilized on a magnetic carrier such as silanized magnetite or on polymeric magnetic (chitin) or magnetizable particles. Alternatively, it is possible to begin with a standard affinity chromatography gel and to modify it magnetically by passing ferrofluid through the gel column [117]. Examples of magnetically assisted separation of enzymes are shown in Table 3.
3.2.2
Electro-Extraction
As both the phases of ATPSs are electrically conductive, application of electric fields in these systems gives rise to electrokinetic mass transfer of charged species. Thus ATPS was shown as a medium for electrophoretic separation with two aqueous phase interfaces providing the stability against convection and facilitating product recovery [126]. Proteins have been directed into either the
Multistage Magnetic and Electrophoretic Extraction of Cells, Particles and Macromolecules |
175 |
top or the bottom phases of PEG/DX system employing 20–50 Vm–1 electric fields perpendicular to the phase interfaces. They could achieve separation of binary mixtures in both batch and continuous modes by operating between isoelectric points and directing oppositely charged proteins into opposite phases.
Recently several studies have been reported regarding improved extractive separation by the application of electric fields to both traditional organic solvent extraction systems and ATPSs [126–130]. Scott and Wham [127] applied an electric field to create emulsion with high interfacial area and contacting with the continuous phase and also to induce coalescence in a novel counter current extractor. Increased mass transfer was achieved due to the altered convection currents within and around the oscillating aqueous drops dispersed in a continuous, nonconducting, organic phase under the influence of a pulsating electric field [129]. Electroextraction with an applied field of 250 Vm–1 was successfully employed to recover citric acid from water using n-butanol (saturated with water) as solvent [131].
Levine and coworkers [132, 133] have reported that electrophoretic transport of proteins across the interface of ATPS is greatly impeded in one direction. They have indicated that the electrophoretic transfer of proteins is readily achieved if the protein is migrating from its less-preferred phase (which is not favorable according to its equilibrium partition behavior) to the morepreferred phase. They also observed that the protein does not migrate in the opposite direction, that is, from the more-preferred phase to the less-preferred phase under similar conditions. However, Theos and Clark [126] have shown that the protein can be made to transfer across the aqueous two-phase boundary in both directions.
Scale-up of ATPE, which is useful in the isolation and purification of bioproducts [134, 135], depends to a large extent upon the rapid demixing of the phases with the desired product concentrated in one of them. Pairs of phases involved in ATPE are characterized by high viscosities, low interfacial tensions, and similar densities [70, 80]. These properties lead to slow demixing rates of these phases which has been counteracted by centrifugation, column contacting, or electrokinetic demixing – each having its own drawbacks [85]. Electrokinetic demixing has been shown to increase demixing rates of ATPSs (up to 100 ml) more than fivefold in a manner that depends on field strength, field polarity, concentration of partitioning anion, and phase composition [91, 92]. Electrokinetic demixing is also potentially useful in situations such as low gravity where an additional force has to be introduced for the demixing of the equilibrated phases [136]. Operation of electrokinetic demixing on a commercial scale requires further understanding of the fundamentals involved in the process, which is the motivation for our recent work where the objective was to identify the mechanism of enhanced demixing [137].
When two polymers are dissolved in aqueous solution at concentrations that cause phase separation, certain dissolved ions such as phosphate are unequally partitioned between the phases [138] leading to an electrical potential across the interface [139] and an apparent electrokinetic potential at the surface of the dispersed phase droplets [140, 141]. As a consequence of the latter, droplets of dispersed phase move in the continuous phase in the presence of an externally
176 |
K.S.M.S. Raghavarao et al. |
applied electric field [141–143]. It is therefore possible to control demixing rates by application of an electric field to ATPS emulsions of appropriate ionic composition. Brooks and Bamberger [136] initially demonstrated enhanced emulsion clearing on a 1-ml scale. They demonstrated qualitatively, by monitoring the system turbidity in a 1-ml chamber, that electrophoretic mobility of the phase droplets enhances the phase demixing.
The effects of the electric field strength, phosphate ion concentration, temperature, field polarity, and phase composition on the demixing rate of PEG/DX system were evaluated quantitatively. It was found that an optimum field strength of around 25 Vm–1 exists at which the demixing is most rapid. In an optimized system this causes a twofold decrease in demixing time relative to that at zero field using normal polarity and a sixfold decrease in demixing time relative to that at zero field in the case of reverse polarity (electric field opposing gravitational settling). Normal polarity refers to anode at the top of the column and reverse polarity refers to anode at the bottom. Paradoxically, both normal and reverse polarity fields increased the demixing rate [91, 92].
Brooks and coworkers [136, 141] measured drop electrophoretic mobilities in ATPSs. They were surprised to discover that the sign of the droplet mobilities was opposite to that predicted from the phosphate partition and the Donnan potential. They also found mobility to be directly proportional to drop radius, which is a contradiction of standard colloid electrokinetic theory [144]. Levine [140] and Brooks et al. [141] hypothesized that a dipole potential at the phase boundary oriented in a way that reverses the potential gradient locally is responsible for the paradox of the sign of electrophoretic mobilities of ATPS droplets.
In our recent study [137], it was confirmed that the droplet electrophoretic mobility increased with increasing drop diameter and the increase was explained based on the electroosmotic flow generated due to the additional internal diffuse double layer of the droplet. These values were compared with the predicted mobilities obtained from the electrophoresis theory. The effect of field strength, polarity, and pH on phase demixing rate was studied. These dependencies were found to be consistent with a model based on the electroosmotic flow:
–u = |
d2 (ÇD – Çc ) g 3hD + 3hc |
dsE E |
|
|
(35) |
|
002 07 |
+ 0003 |
|||||
|
18hc |
3hD + 2hc |
2(3hD + 2hc |
+ sE2 /l |
|
|
and electrophoretic mobility (mE) can be velocity [137] as
mE = – u31E = 0002dsE 2
E 2(3hD + hc + sE /l)
obtained from the electrophoretic
(36)
Raghavarao et al. [91, 92] have also measured the electrophoretic mobilities of the individual phase droplets suspended in the other phase for PEG/DX system using a micro-electrophoresis unit. These values compared well with the predicted mobilities obtained from the electrophoresis theory. Analysis of these
Multistage Magnetic and Electrophoretic Extraction of Cells, Particles and Macromolecules |
177 |
data by the electroosmotic flow model [137] was found useful in resolving two paradoxes:
1.The direction of migration of drops is the opposite of that predicted by colloid electrokinetics.
2.The phase demixing rate increased irrespective of the sign of the applied electric field.
In the case of normal polarity the field supplements the buoyant rise or fall velocities of the phase drops thereby enhancing phase demixing rate over that in the absence of a field. Interestingly, in reverse polarity, though the field works against the buoyancy of the phase drops, it assists their growth in the dispersion zone until buoyancy takes over causing faster phase demixing. As the buoyant velocity of a drop is proportional to the square of its diameter while the electrophoretic velocity is proportional to its diameter, the demixing rate is faster in reverse polarity than in normal polarity.
3.3
Multistage Method
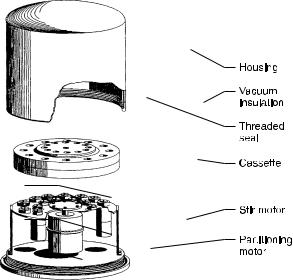
A schematic representation of the ORSEP is shown in Fig. 15. The extractor consists of two disk-shaped plates into which 24 cylindrical cavities have been machined along the space between two concentric circles of the plate faces. When the plates are joined and securely bolted together to form a leak-tight seal, the cavities can be aligned to form an extraction chamber. Access to the chambers is through a small, stoppered port above each cavity of the top plate.
Fig. 15. ORSEP multi-stage countercurrent distribution apparatus
178 |
K.S.M.S. Raghavarao et al. |
In order to exploit surface tension driven phase separation, the top cover plate is fabricated from a hydrophobic plastic (e.g., Plexiglas, Lexan, etc.) while the bottom plate is machined from stainless steel. The entire unit is mounted to a base consisting of 24 miniature stirbar drivers, which are intended to provide adequate mixing by means of a stirbar installed in each chamber.
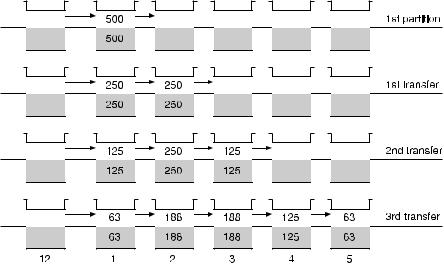
Multi-stage extraction using the ORSEP [95] is intended to mimic the procedure known as ‘Craig extraction’ or counter-current distribution (CCD). As illustrated in Fig. 16, this technique provides a means of isolating and purifying a mixture of solutes by sequentially contacting the top phase of each chamber with the bottom phase from the adjacent chamber, in a manner that mimics counter-current extraction.
The fraction of original solute, added in the first stage, in chamber n after r total transfers is
r!
f (n, r) = 06pn (1 – p)n – r (37) n! (n – r)!
where p = E/(E + 1) and E = KR. The phase volume ratio, R, is defined as the ratio of the top phase volume to the bottom phase volume, and the solute partition coefficient, K, is defined as the ratio of the concentration of solute in the top phase to that in the bottom phase. Multi-stage partitioning of a solute results in a solute concentration profile that closely approaches a Gaussian distribution. Suitable mixing design was provided in a second-generation extractor, ADSEP, for the necessary vertical bulk motion required to achieve adequate momentum transfer between the two fluid phases.
Fig. 16. A schematic representation of Craig extraction
Multistage Magnetic and Electrophoretic Extraction of Cells, Particles and Macromolecules |
179 |
3.4
Theory and Mathematical Models
Counter-current distribution has gradually evolved into a preferred multi-stage contacting method for aqueous two-phase partitioning of biological materials [71]. However, scale-up of this technique beyond the laboratory has been hampered by the lack of a comprehensive model that accounts for the influence of non-idealities on the partitioning process. Counter-current distribution of a mixture of solutes has been modeled as an equilibrium staged process which generates a solute concentration profile that can be represented by a Gaussian error distribution. However, aqueous two-phase CCD is an inherently nonequilibrium process and can result in non-ideal behavior. This is mainly due to factors such as non-alignment of the phase interface and phase cut location, incomplete phase demixing, incomplete solute mass transfer, and perturbation of the phase system composition as a result of solute loading and system dilution. In general, a perturbation of the phase composition in the feed stage due to these and other factors will result in a stage-to-stage variation in the solute partition coefficient (K), the phase volume ratio (R), and the total system volume (VT ). This can translate into a significant deviation from ideal CCD performance. Guinn [95] has developed a comprehensive material balance model for non-ideal CCD and tested it using the ORSEP multi-staged extractor described earlier.
In stage-independent CCD operation K, R, and VT do not vary from stage to stage. However, stage-independent CCD operation is approached only for those limited cases for which solute loading has a negligible impact on phase system composition, total system volume, and phase volume ratio and for which mass transfer and phase demixing have reached equilibrium. Our objective is to begin with the assumption of stage-independent partitioning in order to develop the general equations which describe solute partitioning in CCD but take into account the effect of liquid interface and phase cut nonalignment on CCD performance. The model will then be systematically generalized for nonideal operation by applying an empirical demixing model to account for nonequilibrium phase demixing and a model to account for the effect of solute partitioning and feed stage dilution on the perturbations of the phase volume ratio. Perturbation of the feed stage due to any of these non-equilibrium events will be conveyed as a deviation from ideal CCD operation to each successive stage.
In a CCD apparatus, phase transfer is accomplished by slicing the two-phase system along the plane joining the upper and lower chamber halves. In the unlikely event that the phase interface is aligned with this phase cut plane, Eq. (37) can be applied to predict the solute composition in each phase. A more general treatment takes into account the non-alignment of the liquid interface and phase cut.
Three distinct volume regions are defined as shown in Fig. 17 for two adjacent extraction stages of a typical CCD apparatus containing a total of n extraction stages. Light phase is transferred from the left adjacent cavity (n – 1) to the right cavity (n) by slicing the phase system at the junction of the two

180 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
K.S.M.S. Raghavarao et al. |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fig. 17. CCD extraction model
extractor rings (along the heavy horizontal line in the figure). In general, the liquid-liquid interface may be above or below the phase system cut as indicated by the volume V*. The total liquid volume,VT and the volume of the lower cavity half, Vb , is fixed by the geometry of the extractor.
If xs and ys are the mass concentrations of the solute in the light and heavy phases respectively and LP and HP are the volumes of the light and heavy phases, then the solute balance over stage n following transfer r is
[(VT – Vb – V * ) xs + V * ys ]n –1,r –1 – [(VT – Vb – V * ) xs + V * ys ]n, r –1
[LP xs + HP ys ]n,r – [LP xs + HP ys ]n,r –1
Rearranging, an equation for x in stage n after the r transfer is obtained:
T B |
|
K |
s n –1, r –1 |
||||
V –V – V* |
|
K – 1 |
|
x |
|
|
|
|
71 |
|
|
K(R + 1) s n, r –1 |
|||
T B |
|
|
K |
||||
– V – V |
– V* |
|
K |
– 1 (RK + 1)V |
|||
|
71 |
– 08 x |
|||||
(xs)n, r = 000000001 |
|
(RK + 1)VT |
n, r |
7101 |
|
K(R + 1) |
|
=
(38)
(39)
where the partition coefficient K = xs /ys and the phase volume ratio R = LP /HP . The misalignment volume V* = HP –VB is zero by definition when the liquid interface is at or below the plane of the phase cut. For stage-independent operation, K, VT , and R are constant and Eq. (39) can be rewritten as
T B |
K |
|
s n –1, r –1 |
|
|
|
V –V – V* |
K – 1 |
(x ) |
|
|
|
|
71 |
|
|
|
|||
– V – V |
– V* |
|
K – 1 (RK + 1)VT |
(x ) |
|
|
|
71 |
– 081 |
n, r –1 |
|||
T B |
|
K |
K(R + 1) |
s |
||
(xs)n, r = 0000000543 (40) |
||||||
|
|
(RK + 1)VT |
|
|
||
|
|
7101 |
|
|
||
|
|
K(R + 1) |
|
|
||