

Astruc D. - Modern arene chemistry (2002)(en)
.pdf9.2 {Os(NH3)5}2þ --- The Pentaammineosmium(II) Fragment 313
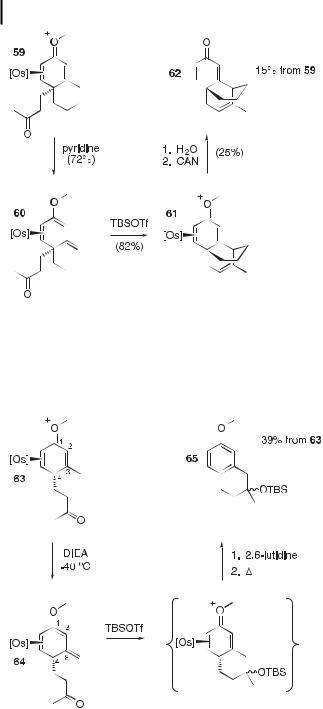
Scheme 5. The synthesis of substituted decalins by means of a Michael–aldol ring-closure sequence.
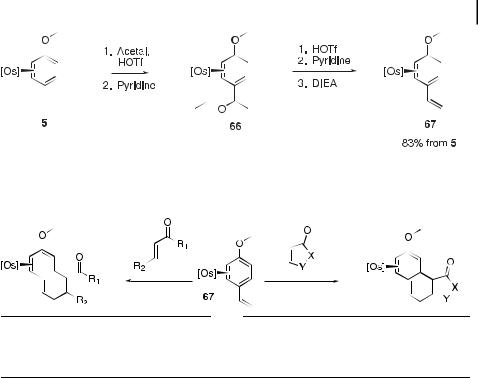
from [Os]-anisole (5) in two steps and 84 % yield (Scheme 8). Acetal substitution at C4 affords 66, from which a net elimination of ethanol generates 67.
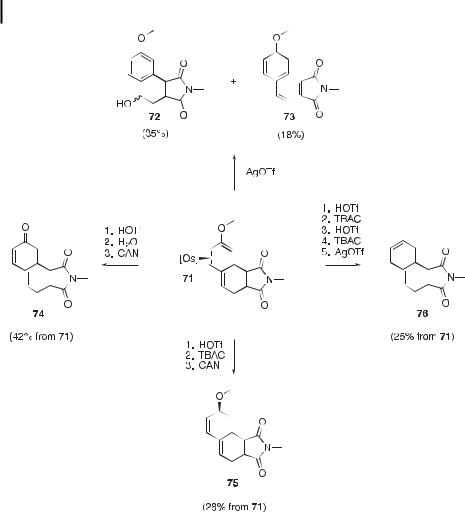
Complex 67 undergoes Diels–Alder reactions with a variety of dienophiles to a ord bicyclic (68 and 69) or tricyclic (70 and 71) vinyl ether complexes (Table 10) [26]. The N- methylmaleimide adduct 71 has been elaborated in a number of ways prior to decomplexation (Scheme 9). Oxidation of 71 with AgOTf produces the re-aromatized species 72 and 73 in yields of 35 % and 18 %, respectively. Oxidation following hydrolysis of the vinyl ether group in 71 generates the enone 74 in 42 % yield. Oxidation following the net reduction of the vinyl ether group in 71 generates the 1,3-diene 75 in 28 % yield. Finally, a series of two hydride reductions performed on 71 generates a diene complex, from which 76 can be liberated in 25 % yield by oxidative decomplexation. The cyclopentenone adduct 70
314 9 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
Scheme 6. The synthesis of a bridged tricyclic system by means of a Michael–aldol ring-closure sequence.
Scheme 7. The synthesis of a tetralin by means of a Michael–aldol ring-closure sequence.
9.2 {Os(NH3)5}2þ --- The Pentaammineosmium(II) Fragment 315
Scheme 8. Synthesis of the 4-methoxystyrene complex 57.
Tab. 10. Diels-Alder reactions with complex 67.
Complex |
R1 |
R2 |
Catalyst |
68 |
H |
H |
LiOTf |
69 |
CH3 |
Ph |
BF3 Et2O |
Complex |
X |
Y |
Catalyst |
|
|
|
|
70 |
CH2 |
CH2 |
* |
71 |
NaCH3 |
CbO |
– |
* Reaction performed in H2O.
(Table 10) may also be elaborated through a hydrolysis sequence to a ord a tricyclic dienone similar to 74 in 40 % yield from 67.
9.2.7
Aniline
Aniline complexes of [Os] are more reactive toward electrophiles than their anisole analogues, presumably because of the less electronegative/more electron-releasing nature of the nitrogen atom. Unfortunately, nucleophilic addition to the resulting anilinium intermediates is di cult and this fact limits the use of this methodology for aniline dearomatization. Nevertheless, [Os]-aniline chemistry has been successfully used in a number of substitution and addition reactions, including an e cient one-pot sequence that generates a di-spiro tetracyclin core through a Michael–Michael–Michael ring-closure.
9.2.7.1 Electrophilic substitution
Aniline complexes are unique in [Os] chemistry in that h2-arene coordination competes with h1-nitrogen coordination. However, h2-coordination becomes favored when the nitrogen is substituted. For example, the N-ethyl aniline complex 77 is isolated solely in its ring-bound form (Table 11). Without Lewis acid promotion, 77 will undergo regioselective addition of
316 9 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
Scheme 9. Elaborations of the complexed Diels–Alder cycloaddition product 71.
Michael acceptors exclusively at C4 to ultimately generate substitution products in high yields [27]. Reaction with acetic anhydride (entry 3) yields the C4-acylated complex. Decomplexed anilines are obtained by heating the substituted complexes at 70 C in a coordinating solvent such as CH3CN. For example, the MVK product (entry 1) can be isolated in 74 % yield [27].
9.2.7.2 4H-Anilinium Michael Additions
As stated above, the isolation of ring-bound [Os]-aniline is di cult because the nitrogenbound form competes with the ring-bound form. However, if the nitrogen is first protected with a TMS group, complexation at the ring proceeds cleanly to yield 78 (Table 12). Deprotection and C4-protonation using HOTf forms the 4H-anilinium complex 79. This com-

9.2 {Os(NH3)5}2þ --- The Pentaammineosmium(II) Fragment 317
Tab. 11. Electrophilic substitution reactions of the N-ethylaniline complex 77.
Entry |
Electrophile |
Solvent(s) |
E |
Yield (%) |
1 |
|
MeOH |
|
94 |
2 |
|
MeOH |
|
89 |
3 |
|
CH3CN/DMSO |
|
87a |
a ¼ Reaction run in the presence of DMAP.
Tab. 12. Michael addition reactions to the 4H-anilinium complex 79.
Entry |
Michael Acceptor |
E |
Yield (%) |
1 |
|
|
83 |
2 |
75 |
3* |
86 |
* ¼ i. DIEA, CH3CN @ 40 C; ii. TBSOTf, CH3CN @ 40 C; iii. H2O.

3189 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
pound will add Michael acceptors (entries 1–3) anti to the face involved in metal coordination and regioselectively at C4 to a ord 4H-anilinium adducts (80) in high yields (Table 12) [27].
9.2.7.3 Electrophilic Addition Reactions
In the presence of Lewis acids, N-substituted aniline complexes of [Os] also add electrophiles at C4, again at the arene face opposite to that involved in metal coordination. This reaction has been shown to be general for a broader range of Michael acceptors than may be utilized with anisole complexes of [Os]. The N-ethyl aniline complex, for example, adds Michael acceptors and acetals in yields ranging from 53–95 % (Table 13, entries 1–6) [27]. The N,N-dimethyl aniline complex (entries 7–9) also adds Michael acceptors to C4 in moderate to high yields (Table 13) and adds to the d-carbon of an a,b,g,d-unsaturated ester (entry 3).
9.2.7.4 Michael–Michael–Michael Ring-Closure
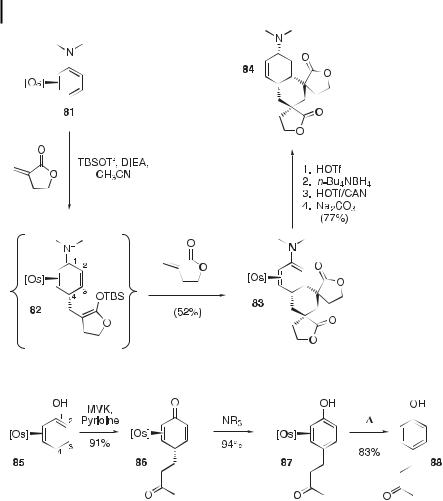
The N,N-dimethyl aniline complex 81 undergoes a Michael–Michael–Michael ring-closure in the presence of excess a-methylene-g-butyrolactone (Scheme 10). In this sequence, the intermediate enolate 82 intercepts another molecule of the Michael acceptor, which then closes at C3 to generate the di-spiro tetracyclic complex 83. The tetracyclic adduct is generated as a single diastereomer, and its structure has been confirmed by X-ray di raction analysis. This molecule undergoes reduction of the enamine through protonation at C2 followed by a stereochemically anomalous hydride reduction at C1. Oxidation of the metal fragment liberates 84 in 77 % yield (based on 83) [28].
9.2.8
Phenol
Phenol complexes of [Os] display pronounced reactivity toward Michael acceptors under very mild conditions. The reactivity is due, in part, to the acidity of the hydroxyl proton, which can be easily removed to generate an extended enolate. Reactions of [Os]-phenol complexes are therefore typically catalyzed using amine bases rather than Lewis acids. The regiochemistry of addition to C4-substituted phenol complexes is dependent upon the reaction conditions. Reactions that proceed under kinetic control typically lead to addition of the electrophile at C4. In reactions that are under thermodynamic control, the electrophile is added at C2. These C2-selective reactions have, in some cases, allowed the isolation of o- quinone methide complexes. As with other [Os] systems, electrophilic additions to phenol complexes occur anti to the face involved in metal coordination.
9.2.8.1 Electrophilic Substitution Reactions
When the parent phenol complex of [Os] 85 is combined with MVK and pyridine in acetonitrile, a Michael addition occurs to give the 4H-phenol complex 86 in 91 % yield (Scheme 11). This complex is remarkably stable and resists rearomatization even when allowed to stand in an acidic solution of acetonitrile for 24 h. However, addition of an amine base induces rearomatization to yield complex 87. This complex may be heated to a ord the demetalated substitution product 88 (raspberry ketone) in 71 % yield (based on 85) [29].

9.2 {Os(NH3)5}2þ --- The Pentaammineosmium(II) Fragment 319
Tab. 13. Scope of electrophilic additions for N-substituted aniline complexes.
Entry |
R1 |
R2 |
Electrophile |
E |
Isomersa |
Yield (%)b |
1 |
H |
Et |
|
|
1:1 |
53 |
2 |
H |
Et |
|
|
– |
82 |
3 |
H |
Et |
|
|
– |
95 |
4 |
H |
Et |
|
|
– |
86c |
5 |
H |
Et |
|
|
– |
81d |
6 |
H |
Et |
|
|
4:1 |
77e |
7 |
Me |
Me |
6:1 |
80 |
8 |
Me |
Me |
2:1 |
63 |
9 |
Me |
Me |
– |
73 |
a Isomer ratio for asymmetric carbon located on R. This ratio is based on the relative heights of 13C NMR resonances and is thus approximate. b Unoptimized yields. c Reaction performed at 40 C.
d ¼ 1. HOTf (1 eq.), 2. H2O. e ¼ 1. HOTf (1 eq.), 2. MeOH.
320 9 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
Scheme 10. A Michael–Michael–Michael ring-closure reaction sequence with complex 81.
Scheme 11. The osmium(II)-promoted C4 alkylation of phenol with MVK.
9.2.8.2 Michael Addition Reactions
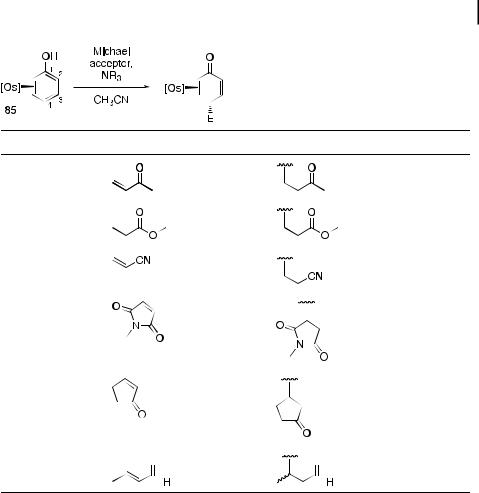
Base-catalyzed The parent phenol complex 85 undergoes a variety of base-catalyzed Michael addition reactions at room temperature to generate 4H-phenol complexes in yields in the range 79–99 % (Table 14) [29]. Michael additions with MVK, methyl acrylate, acrylonitrile, N-methylmaleimide, cyclopentenone, and crotonaldehyde (entries 1–6, respectively) proceed with high regioand stereocontrol. Demetalation and isolation of the dienone is typically accompanied by tautomerization to the aromatic substitution product.
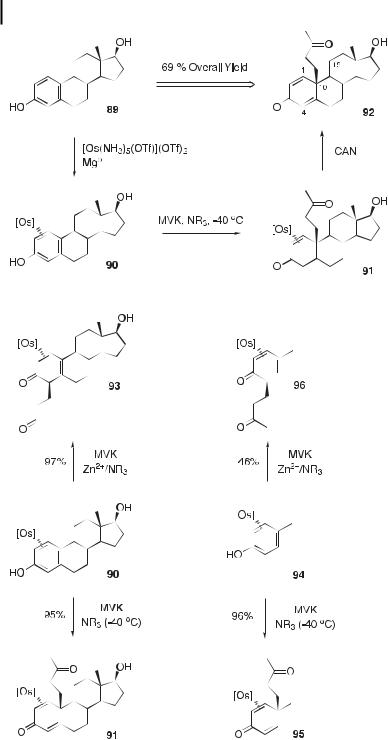
Estradiol A notable application of phenol activation by [Os] is the C10 alkylation of estradiol 89 (Figure 5). The metal fragment preferentially binds to the a face of the steroid (to form 90) and thus promotes the addition of MVK to the b face. This forms a quaternary
9.2 {Os(NH3)5}2þ --- The Pentaammineosmium(II) Fragment 321
Tab. 14. Conjugate addition reactions with the h2-phenol complex 85 and various Michael acceptors.
Entry |
Michael Acceptor |
E |
Yield (%) |
1 |
|
|
91 |
2 |
|
|
81a |
3 |
|
|
99a |
4 |
|
|
98 |
5 |
79 |
6 
 83b
83b
a ¼ Reaction run with Zn(OTf )2 as co-catalyst. b ¼ Reaction run with BF3 instead of NR3.
center at C10 (91), and demetalation releases the steroid 92 in 69 % yield (based on 89). This process results in a steroid that has the C10 stereochemistry of androgens and, consequently, suggests a method for converting estrogen cores to androgen cores in a three-step sequence [30].
Regiocontrol Although Michael additions to [Os]-phenol occur selectively at C4, addition at C2 is thermodynamically favored for phenol complexes that are substituted at C4. For C4substituted phenol complexes, the regiochemistry can be controlled by varying the time, temperature, and catalyst (Figure 6) [29]. Additions of MVK to the estradiol complex 90 and the p-cresol 94 at 40 C in the presence of an amine base catalyst result in regioselective C4 alkylation in high yields (91 and 95). However, when this reaction is performed at room temperature in the presence of a Zn2þ co-catalyst, the Michael acceptor adds at C2 to a ord
322 9 Osmiumand Rhenium-Mediated Dearomatization Reactions with Arenes
Fig. 5. The C(10) alkylation of b-estradiol using osmium(II) to dearomatize the A ring.
Fig. 6. Regiocontrol of the electrophilic addition reaction of MVK to complexes 90 and 94.