Astruc D. - Modern arene chemistry (2002)(en)
.pdf
|
|
|
References |
433 |
|
V. Marvaud, D. Astruc, New J. Chem. |
|
|
|
49 |
64 |
E. M. M. de Brabander-van den Berg, |
||
|
1997, 21, 1309. |
|
E. W. Meijer, Angew. Chem. Int. Ed. Engl. |
|
50 |
V. Sartor, L. Djakovitch, J.-L. Fillaut, |
|
1993, 32, 1308. |
|
|
F. Moulines, F. Neveu, V. Marvaud, J. |
65 |
C. Wo¨rner, R. Mu¨lhaupt, Angew. Chem. |
|
|
Guittard, J.-C. Blais, D. Astruc, J. Am. |
|
Int. Ed. Engl. 1993, 32, 1306. |
|
|
Chem. Soc. 1999, 121, 2929. |
66 |
C. Vale´rio, J. Ruiz, E. Alonso, P. |
|
51 |
a) S. Rigaut, M.-H. Delville, D. Astruc, |
|
Boussaguet, J. Guittard, J.-C. Blais, D. |
|
|
J. Am. Chem. Soc. 1997, 119, 1132; b) S. |
|
Astruc, Bull. Soc. Chim. Fr. 1997, 134, |
|
|
Rigaut, M.-H. Delville, J. Losada, D. |
|
907. |
|
|
Astruc, Inorg. Chim. Acta 2002, 334, 225; |
67 |
F. Moulines, L. Djakovitch, M.-H. |
|
|
c) D. Astruc, in Electron Transfer in |
|
Delville, F. Robert, P. Gouzerh, D. |
|
|
Chemistry (Ed.: V. Balzani), Vol. II, Wiley- |
|
Astruc, J. Chem. Soc., Chem. Commun. |
|
|
VCH, Weinheim, 2001, pp. 714–803. |
|
1995, 463. |
|
52 |
F. Moulines, B. Gloaguen, D. Astruc, |
68 |
F. Moulines, L. Djakovitch, D. Astruc, |
|
|
Angew. Chem. Int. Ed. Engl. 1992, 28, |
|
New J. Chem. 1996, 20, 1071. |
|
|
458. |
69 |
V. Sartor, S. Nlate, J.-L. Fillaut, F. |
|
53 |
G. R. Newkome, X. Lin, J. K. Young, |
|
Djakovitch, F. Moulines, V. Marvaud, |
|
|
Synlett 1992, 53. |
|
F. Neveu, J.-C. Blais, New J. Chem. 2000, |
|
54 |
C. Vale´rio, E. Alonso, J. Ruiz, J.-C. |
|
24, 351. |
|
|
Blais, D. Astruc, Angew. Chem. Int. Ed. |
70 |
S. Nlate, J. Ruiz, D. Astruc, Chem. |
|
|
1999, 38, 1747. |
|
Commun. 2000, 417. |
|
55 |
C. Vale´rio, F. Moulines, J. Ruiz, J.-C. |
71 |
S. Nlate, J. Ruiz, V. Sartor, R. Navarro, |
|
|
Blais, D. Astruc, J. Org. Chem. 2000, 65, |
|
J.-C. Blais, D. Astruc, Chem. Eur. J. 2000, |
|
|
1996. |
|
6, 2544. |
|
56 |
B. Gloaguen, D. Astruc, J. Am. Chem. |
72 |
a) B. Marciniec, in Applied Homogeneous |
|
|
Soc. 1990, 112, 4607. |
|
Catalysis with Organometallic Compounds |
|
57 |
D. Buchholz, B. Gloaguen, J.-L. |
|
(Eds.: B. Cornils, W. A. Herrmann), |
|
|
Fillaut, M. Cotrait, D. Astruc, Chem. |
|
VCH, Weinheim, 1996, Vol. 1, Chap. 2.6; |
|
|
Eur. J. 1995, 1, 374. |
|
b) L. N. Lewis, J. Stein, K. A. Smith, in |
|
58 |
D. Buchholz, D. Astruc, Angew. Chem. |
|
Progress in Organosilicon Chemistry (Eds.: B. |
|
|
Int. Ed. Engl. 1994, 33, 1637. |
|
Marciniec, J. Chojnowski), Gordon and |
|
59 |
F. Moulines, L. Djakovitch, R. Boese, B. |
|
Breach, Langhorne, USA, 1995, p. 263. |
|
|
Gloaguen, W. Thiel, J.-L. Fillaut, M.-H. |
73 |
K. H. Pannel, H. Sharma, |
|
|
Delville, D. Astruc, Angew. Chem. Int. |
|
Organometallics 1991, 10, 954. |
|
|
Ed. Engl. 1993, 105, 1132. |
74 |
P. Jutzi, C. Batz, B. Neumann, H. G. |
|
60 |
a) D. Astruc, C. Vale´rio, J.-L. Fillaut, |
|
Stammler, Angew. Chem. Int. Ed. Engl. |
|
|
J.-R. Hamon, F. Varret, in Magnetism, |
|
1996, 35, 2118. |
|
|
A Supramolecular Function (Ed.: O. |
75 |
J. B. Flanagan, S. Margel, A. J. Bard, |
|
|
Kahn), NATO ASAI Series, Kluwer, |
|
F. C. Anson, J. Am. Chem. Soc. 1978, 100, |
|
|
Dordrecht, 1996, p. 1107; b) C. Vale´rio, |
|
4248. |
|
|
Ph.D. Thesis, Universite´ Bordeaux I, |
76 |
J. Ruiz, D. Astruc, C. R. Acad. Sci. Paris, |
|
|
1996. |
|
t. 1, Se´rie II c, 1998, 21. |
|
61 |
C. Vale´rio, J.-L. Fillaut, J. Ruiz, J. |
77 |
R. L. Collins, J. Chem. Phys. 1965, 42, |
|
|
Guittard, J.-C. Blais, D. Astruc, J. Am. |
|
1072. |
|
|
Chem. Soc. 1997, 119, 2588. |
78 |
S. J. Green, J. J. Pietron, J. J. Stokes, |
|
62 |
a) I. Cuadrado, M. Mora´n, C. M. |
|
M. J. Hostetler, H. Vu, W. P. Wuelfing, |
|
|
Casado, B. Alonso, F. Lobete, B. Garcia, |
|
R. W. Murray, Langmuir 1998, 14, 5612. |
|
|
J. Losada, Organometallics 1996, 15, 5278; |
79 |
C. B. Gorman, J. C. Smith, M. W. |
|
|
b) K. Takada, D. J. Diaz, H. Abrun˜a, I. |
|
Hager, B. L. Parhurst, H. |
|
|
Cuadrado, C. M. Casado, B. Alonso, M. |
|
Sierzputowska-Gracz, C. A. Haney, J. |
|
|
Mora´n, J. Losada, J. Am. Chem. Soc. 1997, |
|
Am. Chem. Soc. 1999, 121, 9958. |
|
|
119, 10763. |
80 |
R. Murray, in Molecular Design of Electrode |
|
63 |
E. Alonso, C. Vale´rio, J. Ruiz, D. |
|
Surfaces (Ed.: R. Murray), Wiley, New |
|
|
Astruc, New J. Chem. 1997, 21, 1119. |
|
York, 1992, p. 1. |
|
434 |
12 Activation of Simple Arenes by the CpFeþ Group |
|
|
|
81 |
P. J. Dandliker, F. Diederich, M. Gross, |
|
Commun. 1999, 1119; B. Dardel, R. |
|
|
|
B. Knobler, A. Louati, E. M. Stanford, |
|
Deschenaux, C.-O. Turrin, J. Chiffre, |
|
|
Angew. Chem. Int. Ed. Engl. 1994, 33, 1739. |
|
D. de Montauzon, J.-C. Daran, A.-M. |
82 |
G. R. Newkome, R. Gu¨ther, C. N. |
|
Caminade, E. Manoury, G. Balavoine, |
|
|
|
Moorefield, F. Cardullo, L. Echegoyen, |
|
J.-P. Majoral, Macromolecules 2000, 33, |
|
|
F. Pe´rez-Cordero, H. Luftmann, Angew. |
|
7328; M. Mosbach, W. Schuhman, |
|
|
Chem. Int. Ed. Engl. 1995, 34, 2023. |
|
Patent No. DE 19917052, 1999, CA 133: |
83 |
H.-F. Chow, I. Y.-K. Chan, D. T. W. |
|
293175; J. Ipatschi, R. Hosseinzadeh, |
|
|
|
Chan, R. W. M. Kwok, Chem. Eur. J. 1996, |
|
P. Schlaf, Angew. Chem. Int. Ed. 1999, |
|
|
2, 1085. |
|
38, 1658; M. Even, E. Serrano, |
84 |
P. J. Dandliker, F. Diederich, J. P. |
|
Macromolecules 1999, 32, 5193; H. C. |
|
|
|
Gisselbrecht, A. Louati, M. Gross, |
|
Yoon, M.-Y. Hong, H.-S. Kim, Anal. |
|
|
Angew. Chem. Int. Ed. Engl. 1996, 34, 2725. |
|
Chem. 2000, 72, 4420; H. C. Yoon, M.-Y. |
85 |
J. Issberner, F. Vo¨gtle, L. De Cola, V. |
|
Hong, H.-S. Kim, Anal. Biochem. 2000, |
|
|
|
Balzani, Chem. Eur. J. 1997, 3, 706. |
|
282, 121; M. Valentini, P. S. Pregosin, |
86 |
C. B. Gorman, B. L. Parkhurst, W. Y. |
|
H. Ruegger, Organometallics 2000, 19, |
|
|
|
Su, K. Y. Chen, J. Am. Chem. Soc. 1997, |
|
2551; T. D. McCarley, C. J. DuBois Jr., |
|
|
119, 1141. |
|
R. L. McCarley, C. M. Cardona, A. E. |
87 |
D. K. Smith, F. Diederich, Chem. Eur. J. |
|
Kaifer, Polym. Prepr. (Am. Chem. Soc., |
|
|
|
1998, 4, 2353. |
|
Div. Polym. Chem.) 2000, 41, 674; C. |
88 |
a) C. Bossard, S. Rigaut, D. Astruc, |
|
Cardona, T. D. McCarley, A. E. Kaifer, |
|
|
|
M.-H. Delville, G. Fe´lix, A. Fe´vrier- |
|
J. Org. Chem. 2000, 65, 1857; R. |
|
|
Bouvier, J. Amiell, S. Flandrois, P. |
|
Deschenaux, Chimia 2001, 55, 139; B. |
|
|
Delhae`s, J. Chem. Soc., Chem. Commun. |
|
Alonso, E. Alonso, D. Astruc, J.-C. |
|
|
1993, 333; b) J. Ruiz, C. Pradet, F. |
|
Blais, L. Djakovitch, J.-L. Fillaut, S. |
|
|
Varret, D. Astruc, Chem. Commun. 2002, |
|
Nlate, F. Moulines, S. Rigaut, J. Ruiz, |
|
|
1108. |
|
V. Sartor, C. Vale´rio, Advances in |
89 |
Redox potentials of the six cathodic |
|
Macromolecules (Ed.: G. Newkome), JAI |
|
|
|
monoelectronic reductions of C60: A. Xie, |
|
Press Inc., Stamford, CT, 2001, Vol. 5, |
|
|
E. Pe´rez-Cordero, L. Echegoyen, J. Am. |
|
pp. 89–127. |
|
|
Chem. Soc. 1992, 114, 3978. |
91 |
a) Y. Wang, C. Cardona, A. E. Kaifer, J. |
90 |
a) For reports on other ferrocene |
|
Am. Chem. Soc. 1999, 121, 9756; b) A. E. |
|
|
|
dendrimers, see ref. [9], which reviews |
|
Kaifer, M. Gomez-Kaifer, Supramolecular |
|
|
dendrimers of the Madrid group, and: S. |
|
Electrochemistry, Wiley-VCH, Weinheim, |
|
|
Achar, C. E. Immoos, M. G. Hill, V. J. |
|
1999. |
|
|
Catalano, Inorg. Chem. 1997, 36, 2314; R. |
92 |
C. B. Gorman, Adv. Mater. 1997, 9, 1117. |
|
|
Deschenaux, E. Serrano, A.-M. Levelut, |
93 |
R. M. Crooks, M. Zhao, L. Sun, V. |
|
|
Chem. Commun. 1997, 1577; C.-F. Shu, |
|
Chechik, L. K. Yeung, Acc. Chem. Res. |
|
|
H.-M. Shen, J. Mater. Chem. 1997, 7, 47; |
|
2001, 34, 181. |
|
|
C. M. Cardona, A. E. Kaifer, J. Am. |
94 |
Y. Xia, J. A. Rogers, K. E. Paul, G. M. |
|
|
Chem. Soc. 1998, 120, 4023; C. Ko¨llner, |
|
Whitesides, Chem. Rev. 1999, 99, |
|
|
B. Pugin, A. Togni, J. Am. Chem. Soc. |
|
1823. |
|
|
1998, 120, 10274; b) R. Schneider, C. |
95 |
a) S. Marcen, M. V. Jimenez, I. T. |
|
|
Kollner, I. Weber, A. Togni, Chem. |
|
Dobrinovich, F. J. Lahoz, L. A. Oro, J. |
|
|
Commun. 1999, 2415; c) E. G. Oosterom, |
|
Ruiz, D. Astruc, Organometallics 2002, 21, |
|
|
R. J. van Haaren, J. N. H. Reek, P. C. J. |
|
326; b) V. Martinez, J.-C. Blais, D. |
|
|
Kamer, P. W. N. M. van Leeuwen, Chem. |
|
Astruc, Org. Lett. 2002, 4, 651. |

435
13
Charge-Transfer E ects on Arene Structure and Reactivity
Sergiy V. Rosokha and Jay K. Kochi
Abstract
Arenes spontaneously form intermolecular 1:1 complexes with a wide variety of electrophiles, cations, acids, and oxidants that are all su ciently electron-poor to be classified as electron acceptors. Spectral, structural, and thermodynamic properties of these donor/ acceptor associates are described within the context of the Mulliken charge-transfer (CT) formulation. The quantitative analyses of such CT complexes provide the mechanistic basis for understanding arene reactivity in di erent thermal and photochemical processes.
13.1
Introduction
Arenes have historically played a pivotal role in the development of modern organic chemistry, in large part stemming from their p-electronic structures comprised of cyclic arrays of conjugated double bonds. The latter are especially critical to the facile formation of molecular (1:1) complexes with a wide variety of rather common reagents, such as Brønsted and Lewis acids, organic and inorganic cations and oxidants, molecular and ionic electrophiles, etc., all of which are readily recognized as being relatively electron-poor or -deficient, and are thus hereinafter referred to generally as electron acceptors [1–4]. Thermodynamically, such intermolecular complexes of arenes (ArH) and electron acceptors (A) run the gamut from extremely stable and readily isolable in crystalline form to highly labile with lifetimes on a par with molecular collisions [5]. From a mechanistic point of view, these interactions are tantamount to the e cient organization of the reactant arene and the electron acceptor within the prereactive complex [ArH, A] prior to (bimolecular) reaction. For example, in biochemical systems, such assemblies (derived from multiple weak intermolecular interactions) can be dominant, as in enzyme/substrate complexes, antigen/antibody binding, etc., for the variety of regioand stereospecific transformations [6]. At the other extreme, intermolecular interactions of chromophoric (aromatic) functionalities play an important role in the design of nanomolecular devices such as electronic switches, sensors, etc. [7, 8].
Owing to the inextensible range of complex stabilities extant with various ArH/A combinations, it is important to develop an analytical methodology that is universally applicable and capable of identifying the intermolecular interactions accurately and reliably. As such, in
Modern Arene Chemistry. Edited by Didier Astruc
Copyright 8 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30489-4

43613 Charge-Transfer Effects on Arene Structure and Reactivity
this chapter we initially explore how the electronic transitions specifically arising from intermolecular interactions of arenes with various electron acceptors provide a comprehensive basis for intermolecular complex formation according to Mulliken [9]. The quantitative treatment of the static properties of intermolecular arene complexes is then followed by the mechanistic analysis of aromatic reactivity that proceeds from Mulliken’s formulations of intermolecular complexes as dynamic precursors to electron transfer based on Marcus–Hush theory [10, 11].
In Section 13.2, a short presentation of the Mulliken formulation provides the theoretical background for understanding the spectral and thermodynamic characteristics as well as charge distributions in intermolecular complexes. Section 13.3 presents the general structural features of intermolecular complexes and focuses more specifically on the structural changes in the arene donor upon complexation. Such changes lead to arene activation (critical to organometallic catalysis), which is described in Section 13.4. Finally, Section 13.5 illustrates how the recognition of intermolecular complexes as crucial intermediates creates the mechanistic basis for understanding arene reactivity in di erent thermal and photochemical processes. As a result, we emphasize how this unified approach can lead to spectral, structural, and thermodynamic properties, and consequently the reactivity of a wide variety of arene associates, from very weakly bonded encounter complexes to stable organometallic derivatives.
13.2
Mulliken’s Quantitative Description of Intermolecular (Charge-Transfer) Complexes
According to Mulliken [9], arenes are classified as electronic donors (D) in measure with their degree of electron-rich character, as evaluated by their ionization potential (IP, gas phase) or oxidation potential (E ox, solution) [12]. The intermolecular interaction of the arene donor (D) and electronic acceptor (A) spontaneously leads to the electron donor/ acceptor or EDA complex, i.e.
KCT |
ð1Þ |
A þ D ! ½D; A& |
13.2.1
Short Theoretical Background
Intermolecular complexes are generated by the linear combination of the principal van der Waals (cD; A) and dative (cDþA ) states, so that the ground-state (CGS) and excited-state (EES) wavefunctions in the two-state or charge-transfer (CT) model can be expressed as [13]:
CGS |
¼ |
acD; A |
þ |
bcDþA |
ð Þ |
|
|
2 |
|||
CES |
¼ |
bcD; A |
|
acDþA |
ð Þ |
|
|
3 |
The energies of the ground (EGS) and excited (EES) states that are obtained by solving the secular determinant by the variation method (and constraining the mixing coe cients to the normalized a2 þ b2 ¼ 1) can be expressed as [13]:
|
|
|
|
|
13.2 |
Mulliken’s Quantitative Description of Intermolecular (Charge-Transfer) Complexes |
437 |
||||||||||||||
EGS |
¼ ð |
ED; A |
þ |
EDþA |
Þ |
=2 |
ð |
DDA |
2 |
þ |
4HDA |
2 |
Þ |
1=2=2 |
4 |
|
|||||
|
|||||||||||||||||||||
|
|
|
|
|
|
|
|
|
|
ð Þ |
|
||||||||||
EES |
¼ ð |
ED; A |
þ |
EDþA |
Þ |
=2 |
þ ð |
DDA |
2 |
þ |
4HDA |
2 |
Þ |
1=2=2 |
5 |
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
ð Þ |
|
|||||||
and the product of the mixing coe cients is:
jabj ¼ jHDAj=ðEES EGSÞ |
ð6Þ |
Ð |
Ð |
The Coulomb integrals cD; AHcD; A and |
cDþA HcDþA represent the energies ED; A |
and EDþA of the van der Waals and dative states, respectively. The energy gap is DDA ¼
EDþA |
|
ED; A and the electronic coupling matrix element is given by the resonance integral |
Ð |
|
cD; AHcDþA ¼ HDA.
The optical or charge-transfer transition (hnCT) stems from electron promotion from the ground to the excited state of the complex, and it follows from Eqs. (4) and (5), that
hnCT ¼ EES EGS ¼ ðDDA 2 þ 4HDA 2Þ1=2 ð7Þ
Most importantly, the spectral characteristics of the charge-transfer (CT) absorption band also provide a quantitative measure of the electronic coupling element in the intermolecular complex, i.e. [14–16]:
HDA ¼ 0:0206ðnmaxDn1=2emaxÞ1=2=r |
ð8Þ |
where nmax and Dn1=2 are the maximum and the width (in cm 1), respectively, of the chargetransfer band, emax is the extinction coe cient (m 1cm 1), and r is the donor/acceptor separation (A˚ ) in the complex [17].
While the Mulliken description above is based on the valence-bond formalism, complex formation can alternatively be presented in (equivalent) molecular orbital or MO terms [18]. Thus, charge-transfer bonding and antibonding orbitals are formed as the result of HOMO/ LUMO interactions of the donor and acceptor orbitals, and the mixing coe cients refer to the relative participation of donor and acceptor orbitals in the complex wavefunction. Accordingly, the energies of the initial and final (diabatic) states are substituted by the corre-
ÐÐ
sponding Coulomb integrals cDHcD and cAHcA, which represent the electron energies
of the donor and acceptor orbitals, respectively, and the coupling element HDA is given by
Ð
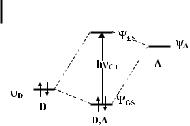
the resonance (exchange) integral HAD ¼ cDHcA, representing the electronic interaction energy in the donor/acceptor dyad. Although the MO approach is less suitable for the consideration of reaction dynamics [19], it often allows a clearer description of the ground-state and optical properties of the intermolecular (charge-transfer) complex, as readily depicted by the simple energy (orbital) diagram in Chart 1, and it will also be employed (interchangeably) hereafter, as appropriate.
13.2.2
Quantitative Evaluation of Arenes as Electron Donors
Donor/acceptor interactions in charge-transfer (CT) complexes are determined primarily by the symmetry and the energetics of the frontier orbitals (HOMO and LUMO) [20]. A
438 13 Charge-Transfer Effects on Arene Structure and Reactivity
Chart 1
quantitative classification of arene ligands according to their electron-donor and -acceptor strengths is generally based in the gas phase on the (vertical) ionization potential (IP) and the electron a nity (EA), respectively. In solution, the appropriate measure is based on the oxidation (E ox) and reduction (E red) potentials. As such, IP and E ox relate to the energetics of the conversion of an arene (ArH) to its cation radical (ArHþ), whereas EA and E ored relate to the formation of the acceptor anion radical (A ). Owing to the high reactivities of the ion radicals of most aromatic compounds, the reversible oxidation and reduction potentials are not generally accessible. However, this problem is partially alleviated by the electrochemical measurement of the irreversible anodic and cathodic waves, Ep a and Ep c, respectively, which are readily obtained from linear sweep or cyclic voltammograms (CV) [21]. Since the values of Ep a and Ep c contain contributions from kinetic terms (E o ¼ bEp þ constant), any comparison with the values of the E o is necessarily restricted to a series of structurally related donors [22].
An alternative measure of the electron-donor properties is the ionization potential (IP), which is experimentally determined from photoelectron spectra in the gas phase. The values of IP di er from the oxidation potential E oox by (cation) solvation, DGs (i.e. E oox GIP þ DGs) [22]. When the variations in DGs are minor, the values of IP such as those listed in Table 1 can be adequate measures of the electron-donor properties of organic compounds applicable to a particular solvent. This generalization is especially tenable for a series of related compounds [22]. Nonetheless, the global plot of the extensive data [4] indicates that the electrondonor property (i.e. HOMO energy) is consistently revealed in the trend of the gas-phase (ionization) potentials relative to the solution-phase oxidation potentials, despite large di erences in structural types. Such a correlation suggests that a similar conclusion pertains to the electron-donor properties of organic anions, for which gas-phase potentials are more di cult to measure consistently. Although the same limitations apply to the use of Ep c as those described above for the anodic counterpart, the general trend shows that gas-phase electron a nities also generally reflect the trend in the reduction potentials measured in solution (i.e. E ored GEA þ DGs) for the large variety of acceptors included in Table 2 [4]. Because of their use as photochemical quenchers, the enhanced values of the reduction potential for the excited singlet and triplet acceptor species A are also listed in Table 2 [4].
13.2.3
Spectral (UV/vis) Probe for the Formation of CT Complexes
The most characteristic spectroscopic feature of CT complexes is a new absorption band in their electronic (UV/vis/NIR) spectra that is not seen in the individual spectra of either the

13.2 Mulliken’s Quantitative Description of Intermolecular (Charge-Transfer) Complexes 439
Tab. 1. Oxidation potentials and ionization potentials of various classes of arenesa
Donorb |
E oox |
IP |
Donorb |
E oox |
IP |
Methylbenzenes |
|
|
Methoxy(hydroxy)benzenes |
|
|
Benzene (BEN) |
2.62 |
9.24 |
Phenol |
1.04 |
8.50 |
Toluene (TOL) |
2.25 |
8.82 |
Anisole |
1.76 |
8.22 |
o-Xylene (o-XY) |
2.16 |
8.56 |
(MA)c |
1.16 |
|
Mesitylene (MES) |
2.11 |
8.39 |
(EA)c |
1.30 |
|
Durene (DUR) |
1.84 |
8.03 |
Polycyclic aromatic hydrocarbons |
|
|
Pentamethylbenzene (PMB) |
1.71 |
7.92 |
Naphthalene (NAP) |
1.54 |
8.14 |
Hexamethylbenzene (HMB) |
1.62 |
7.85 |
Anthracene (ANT) |
1.09 |
7.45 |
(TMT)c |
1.50 |
|
(DMT)c |
1.43 |
|
(TET)c |
1.55 |
|
(OMN)c |
1.34 |
|
in V vs SCE, IP in eV, data from ref. [4].
b The acronym in parentheses will be used hereinafter. c Data from ref. [28], see structure below.
uncomplexed donor or the acceptor. Such absorption bands are universally observed for arene associates with organic and inorganic acceptors, for both weak as well as strong molecular complexes, and even in the case of short-lived collision complexes with KCT f1 [3, 4, 23, 24].
As is apparent from Eq. (7), the energy of the electronic transition in CT complexes is determined as the sum of the two terms, the di erence in the energy of the diabatic states (DDA) and the electronic coupling element (HDA). Depending on their relative values, there can be two limiting cases represented by weak complexes with HDA fDDA and strong complexes with DDA fHDA.
In a weak complex, the wavefunction CGS for the ground state basically describes a nobond (or initial) state (D, A), in which the (final) charge-transfer state (Dþ, A ) does not play
Tab. 2. Reduction potential and electron a nity of (ground-state and excited) arene acceptorsa
Ground-State Acceptor |
E ored |
EA |
Excited Acceptorb |
E ored |
2,6,9,10-Tetracyanoanthracene |
0.45 |
|
2,6,9,10-Tetracyanoanthracene (s) |
2.82 |
9,10-Dicyanoanthracene |
0.98 |
|
9,10-Dicyanoanthracene (s) |
2.88 |
1,2,4,5-Tetracyanobenzene |
0.65 |
1.6 |
1,2,4,5-Tetracyanobenzene (s) |
3.83 |
1,4-Dinitrobenzene |
0.69 |
2.00 |
1.4-Dinitrobenzene (t) |
2.6 |
Picric acid |
0.51 |
|
2-Phenylpyrrolinium (s) |
2.9 |
a E ored in V vs. SCE, EA in eV, data from ref. [4]. b Singlet (s) and triplet (t) states.
440 13 Charge-Transfer Effects on Arene Structure and Reactivity
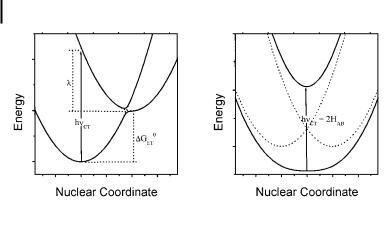
Fig. 1. Optical transitions in charge-transfer complexes in the weak (A) and strong (B) interaction limits.
a significant role, i.e. with mixing coe cients b fa. The ratios of the mixing coe cients a/b for the no-bond and dative functions are inverted in the excited state. According to Mulliken [9], the optical CT transition (hnCT) derives from an intracomplex transfer of a single electron from the donor to the acceptor (see Chart 1). As such, the photoexcitation of the CT absorp-
tion band |
of the electrically neutral EDA complex generates the ion-radical pair [25]: |
hnCT |
½Dþ; A &, where the asterisk denotes an electronically and vibrationally excited |
½D; A& ! |
system. The donor and acceptor components in the Franck–Condon (FC) excited state have the same nuclear coordinates as those in the ground state, and any nuclear relaxation (bond length and angle changes, solvent reorganization, etc.) generates a vibrationally equilibrated excited state. In accordance with Eq. (7), when HDA fDDA, the energy of the transition is determined primarily by the energy di erence of the diabatic states, DDA [26], and hnCT ADDA ¼ DGET þ l, as illustrated in Figure 1.
The energy di erence DGET between the vibronically equilibrated reactant and product states can be considered as the di erence between the IP of the donor and the EA of the acceptor (in the gas phase) or between the corresponding electrochemical potentials (in solution). For a set of structurally related arene donors in the same solvent, a linear (Mulliken) correlation is usually observed experimentally between the donor strength and the CT energy
(due to the relatively small changes in l), i.e.: |
|
Mulliken correlation: hnCT ¼ IP EA þ constant |
ð9Þ |
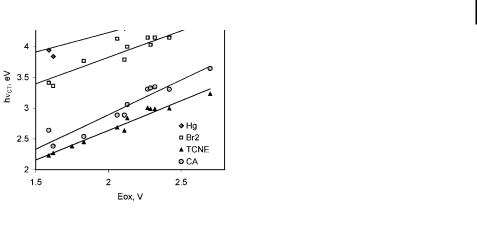
Eq. (9) is applicable when a common acceptor is employed [or vice versa for the electron affinities (EA) of a series of acceptors interacting with a common donor], as predicted by Mulliken [9] and as illustrated in Figure 2.
With increasing electronic interaction between the donor and the acceptor, HDA plays a more important role than DDA in determining the energy of the CT transition. Such e ects are manifested in a deviation of the Mulliken slope from linearity when donors with a wide range of IPs are considered [23b]. (If donors in a narrower IP range are taken, this e ect is
13.2 Mulliken’s Quantitative Description of Intermolecular (Charge-Transfer) Complexes 441
Fig. 2. Mulliken plot for CT complexes of series alkylbenzene donors with di erent acceptors (acceptors: Hg ¼ Hg(O2CCF3)2,
Br2 ¼ bromine, TCNE ¼ tetracyanoethylene, CA ¼ chloroanil). Data from refs. [23a, 23g].
seen as a decrease of the Mulliken slope with increasing values of HDA or acceptor strength [18]). An increased electronic coupling interaction also results in a corresponding increase in the contribution of the dative wavefunction in the CT ground state. Therefore, the mixing coe cient b2 approaches a2, especially for those complexes in which the di erence in the redox potentials of the donor and the acceptor is small. Thus, the extent of electron transfer from the donor to the acceptor (which is equal to the di erence a2 b2) during the photoexcitation becomes smaller. Finally, for strong complexes close to the isergonic point, the absorption is better described as a bonding/antibonding (MO) rather than as a chargetransfer transition.
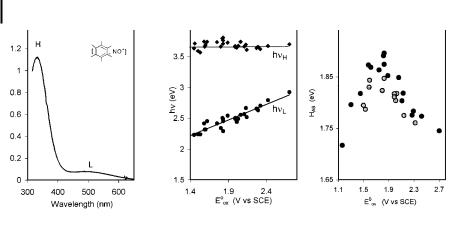
The spectral behavior of strong CT complexes is illustrated by the interaction of the NOþ cation with a series of arenes. The UV/vis electronic spectra consist of two bands [27, 28], which are assigned to optical transitions from the bonding orbital (high-energy band, hnH) and from the arene HOMO (low-energy band, hnL) to the antibonding orbital. From the relationship of hnH to the sum of DDA and HDA in Eq. (7), it is possible to calculate the electronic coupling elements for a number of complexed arenes (based on hnH and the di erence of the arene and nitrosonium potentials EArH 0 ENO 0, taken as DDA). Thus, Figure 3C presents a plot of these high HDA values as a function of Eox 0, with a maximum observed near the isergonic point: EArH 0 AENO 0 ¼ 1:48 vs. SCE.
As EArH 0 approaches the isergonic point, the value of 4HDA 2 parallels DDA 2, and their sum remains more or less constant, thus accounting for the invariance of hnL with EArH 0. In contrast, the low-energy band (hnL) corresponds to the transition from the non-interacting
the antibonding MO, and its energy can be written as [28]: hn |
L ¼ |
|||
arene orbital to1=2 |
=2 þ DDA=2. Since the term ðDDA 2 þ 4HDA 2Þ |
1=2 |
|
|
ðDDA 2 þ 4HDA 2Þ |
|
is nearly constant |
for |
|
di erent arenes, the changes in hnL are mainly determined by the DDA=2 term. In other words, hnL decreases linearly with decreasing values of EArH 0 (Figure 3B).