

Astruc D. - Modern arene chemistry (2002)(en)
.pdf13.2 Mulliken’s Quantitative Description of Intermolecular (Charge-Transfer) Complexes 443
Fig. 4. Correlation of the experimentally determined values of the degrees of charge-transfer (Z) in nitrosonium/arene complexes with the calculated mixing coe cients cb 2 based on HDA and DDA. Data from ref. [28].
ion, the changes in nNO represent an ambiguous (experimental) measure of the degree of charge transfer (designated as Z) from the aromatic donor to NOþ [27]:
Z |
¼ ð |
nNOþ |
2 |
|
nC 2 |
= nNOþ 2 |
|
nNO |
2 |
Þ |
11 |
|
|
|
|
Þ ð |
|
|
ð Þ |
where the subscript NOþ represents the (uncomplexed) cation, C is the complex, and NO the (completely) reduced nitric oxide. Theoretically, the degree of charge transfer can be viewed as the excess charge residing on the NOþ moiety in the CT complex, 2b2 [28]. The calculated values of 2b2 based on HDA and DDA (obtained from UV/vis and electrochemical data) [28] are plotted against the experimental Z values in Figure 4, and the linear correlation confirms the validity of the CT formalism in correctly predicting the changes in the degree of charge transfer with the arene donor strength.
13.2.5
Thermodynamics of Charge-Transfer Complexation
One of the principal measures of donor/acceptor binding in CT complexes is the formation constant, KCT, which is related to the free energy of complex formation DG CT ¼2:3RT logðKCTÞ. The formation constant is usually determined by absorption spectroscopy, according to the commonly utilized Benesi–Hildebrand treatment [30]. The value of KCT is quantitatively evaluated from the graphical plot of the absorbance change ðACTÞ as the donor is progressively added to a solution of the acceptor (or vice versa). Relatively strong binding is typically indicated by experimental values of KCT > 10 m 1. When KCT lies in the range 1 < KCT < 10 m 1, the complex is considered to be weak. Finally, at the limit of very weak donor/acceptor associations with KCT f1, the lifetime of the complex can be of the order of that of a molecular collision, and these are referred to as contact CT complexes [24].
The free-energy changes that accompany CT complex formation, DG CT, include several factors, such as electrostatic interactions, entropy changes, etc. In terms of the CT formalism, the energy gain upon complex formation arises from the energy di erences of the non-
444 13 Charge-Transfer Effects on Arene Structure and Reactivity
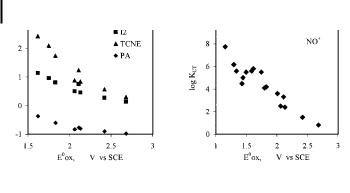
Fig. 5. Typical dependence of the formation constants of arene complexes with di erent acceptors on the oxidation potential of the arene. Data from refs. [23, 28]. I2 ¼ iodine, TCNE ¼ tetracyamethylene; PA ¼ picric acid.
interacting donor and acceptor orbitals, and that in the complex and can be expressed as:
DEDA ¼ ðDDA 2 þ 4HDA 2Þ1=2=2 DDA=2 |
ð12Þ |
Eq. (12) allows changes in the stability of CT complexes consisting of a single acceptor with a series of similar donors (or vice versa) to be evaluated, since the other factors (e.g. entropy, solvation, etc.), are more or less constant for related aromatic compounds. Eq. (12) can be approximated as DE ¼ HDA 2=DDA when DDA 2 gHDA 2 for strongly endergonic and/or weakly interacting systems. Arene complexes with bromine and iodine are examples of such systems [23]. Thus, an increase in the donor ability of the arene results in a decrease in DDA and an increase in DEDA, and a corresponding increase in the formation constants with increasing donor ability of the alkylbenzenes (Figure 5). Similar dependences are observed for arene complexes with other acceptors, as seen in the free energies of complex formation for the CT complexes of di erent alkylbenzenes with TCNE, chloranil, ICl, and 1,3,5- trinitrobenzene, which are linearly related to those of iodine [23b]. The presence of bulky substituents on the benzene ring, as in encumbered arenes, results in a substantial decrease in the formation constant of the CT complex (Table 3) [31]. As such, steric hindrance inhibits the close approach of the donor to the acceptor to ensure e ective orbital interaction, and results in lower HDA values. As can be seen in Table 3, the energy of the CT bands in each series remains roughly constant, which implies that the corresponding DDA values are also constant. On the other hand, the HDA value for the complex with mesitylene can be estimated to be @0.4 eV (based on spectral data from Table 3 and r ¼ 3:5 A˚ [31]), while for the monoand di-tert-butyl analogues, HDA is less than 0.25 eV and 0.15 eV, respectively. In other words, steric hindrance (diminishing HDA) is a dominant factor relative to donor strength in determining the changes in DDA. As a result, the values of KCT for complex formation of chloranil with mesitylene and durene are substantially higher than that with the more electron-rich hexaethylbenzene [31].
In the limit of strongly coupled complexes close to the isergonic region, DDA 2 f4HDA 2, and this approximation leads to the qualitative conclusion that DEDA and HDA are strongly
13.3 Structural Features of Arene Charge-Transfer Complexes 445
Tab. 3. The e ect of steric hindrance on the formation constant for the EDA complexes of chloranil (CA) with alkyl-substituted benzenesa
Arene |
E ox , |
KCT , |
hnCT , |
eCT , |
Arene |
E ox , |
KCT , |
hnCT , |
eCT , |
donor |
V vs SCE |
MC1 |
eV |
MC1cmC1 |
donor |
V vs SCE |
MC1 |
eV |
MC1cmC1 |
MES |
2.11 |
0.80 |
2.87 |
1500 |
DUR |
1.83 |
1.40 |
2.61 |
1600 |
TXYc |
2.09 |
0.29 |
2.90 |
650 |
TMAc |
1.84 |
0.36 |
2.62 |
300 |
DTTc |
2.08 |
0.18 |
2.88 |
240 |
OMAc |
1.84 |
<0.01 |
b |
b |
TTBc |
2.10 |
<0.01 |
b |
b |
DMAc |
1.51 |
<0.01 |
b |
b |
a Data from ref. [35]. b No new bands.
c See structure below.
coupled since the stabilization energy of complex formation is largely determined by the
Ð
donor/acceptor electronic interaction energy HDA ¼ CDHCA. Such a conclusion predicts that the electronic exchange (HDA) between the donor and acceptor orbitals in the CT complex should play a major role in the experimental free-energy change (DGCT), and this is confirmed experimentally by the close relationship between the relative stability of the CT complexes and the electronic interaction term HDA in the case of strongly coupled [ArH, NOþ] complexes [28]. Furthermore, the changes in DGCT in such a series are determined by the second term, DDA=2, since the first term in Eq. (12) is nearly constant for NOþ complexes with the series of alkylbenzenes. This explains the experimental observation of the linear dependency of DGCT on E ored [28], which is qualitatively similar to that observed for weak complexes (Figure 5).
Consideration of the spectral and thermodynamic properties of arene CT complexes thus indicates that they are reasonably described within the framework of recent developments of the Mulliken formalism, in the case of both weak and strong complexes in the highly endergonic and nearly isergonic regions. Accordingly, let us now consider the structural consequences attendant upon charge transfer from the donor to the acceptor in such complexes.
13.3
Structural Features of Arene Charge-Transfer Complexes
The donor/acceptor properties and the electronic coupling interactions determine the redistribution of electron density between the aromatic donor and the electron acceptor upon complexation. Significant changes in structure and reactivity of the coordinated arene can be rationalized in terms of spectral and thermodynamic properties within the framework of the CT formalism. This section is devoted to a consideration of the structural e ects of arene coordination (in terms of donor/acceptor bond distance and type of bonding, distortion of arene planarity, expansion of the aromatic ring, and p-bond localization).

44613 Charge-Transfer Effects on Arene Structure and Reactivity
13.3.1
Bonding Distance of the Donor/Acceptor Dyad in Arene Complexes
The bond distance is the most commonly used measure to gauge the strength of intraor intermolecular bonds between two nuclei. In the case of intermolecular CT complexes, the donor/acceptor separation is determined by the balance between the HOMO-LUMO (bonding) electronic interaction and the repulsion of filled orbitals. Strong CT interactions can result in significant bond shortening relative to the separation determined by the purely van der Waals contact. For example, the interplanar distance of d ¼ 3:42 A˚ between hexamethylbenzene and chloranil in their mixed-stack CT crystals is shortened by 0.2 A˚ compared to the intermolecular distance d ¼ 3:62 A˚ in pure crystals of hexamethylbenzene [2]. The intermolecular separation of bromine from the ring plane of benzene in the 1:1 complex
is 0.5 A˚ shorter than that predicted from the van der Waals radii [32, 33]. Even greater shortening (of about 1 A˚ ) of the donor/acceptor separation relative to the sum of the van der Waals radii is observed in strongly coupled complexes of alkyl-substituted benzenes with NOþ, in which the distance between the nitrogen atom and the aromatic ring is @2.1 A˚ [28].
In di erent types of complexes, the shortening of the donor/acceptor separation with increasing donor ability of the arene (due to enhancement of the HDA=DDA ratio) determines the gain in energy upon complex formation. Thus, the distance between gallium and the aromatic ring in [ArH, Gaþ] complexes varies from d ¼ 2:76 A˚ for benzene to d ¼ 2:42 A˚ for the hexamethylbenzene complex. A similar trend is observed in p-complexes of transition metals. For example, in the 1:1 complexes of arenes with niobium(0), the Nb/ArH distance is decreased by 0.01 A˚ from d ¼ 1:860 A˚ in the toluene to d ¼ 1:850 A˚ in the mesitylene complex, as the arene donor strength is increased. Similar e ects are observed when arene complexes with metals of di erent acceptor strengths are compared. For example, the distance between the arene ligand and the chromium center is reduced by 0.015 A˚ in the benzene complex [(BEN)2CrI]Br (d ¼ 1:597 A˚ ), as compared to the corresponding [(BEN)2Cr0] complexes with d ¼ 1:612 A˚ [2].
The balance between van der Waals repulsion and electronic coupling depends on the stereochemistry. Thus, in complexes of chloranil and TCNE with encumbered hexaalkylsubstituted benzenes (such as hexaethylbenzene and its multiply annulated analogues), the interplanar distance is larger [31], and HDA and KCT are lower, than those in the corresponding complexes with mesitylene and durene, in spite of the weaker donor ability of the latter. On the other hand, sterically encumbered donors with van der Waals cavities of su - cient size can allow small acceptors such as the NOþ cation to closely approach the benzenoid ring. The distance between the nitrogen atom and the arene ring in nitrosonium complexes with hexaethylbenzene (2.08 A˚ ) is nearly the same as that in the hexamethylbenzene complex (2.09 A˚ ) [28], and values of HDA and KCT for the complexes with hindered donors are only slightly lower than those for complexes with relatively unhindered analogues [28].
Organic ligands in transition metal complexes frequently penetrate the coordination sphere of the d-metal to such an extent that the ligand-to-ligand distance is less than the sum of the van der Waals radii. In such cases, steric repulsion between ligands can overcompensate the distance-shortening e ects of charge transfer, and eventually cause an increase in the arene–metal distance as the donicity of the arene ligand increases. For exam-
13.3 Structural Features of Arene Charge-Transfer Complexes 447
ple, the Ru2þ –arene distance in the bis(benzene) complex (1.713 A˚ ) is shorter than that in the corresponding bis(mesitylene) complex (d ¼ 1:726 A˚ ) [2].
13.3.2
Relationship Between Hapticity and Charge Transfer in Arene Complexes
Topological classification of metal–arene complexes in terms of hapticity was originally proposed by Cotton [35] as the basis for distinguishing organometallic complexes and their chemistry, but it was never intended to characterize the chemical bonding between the metal and the ligand. As a ligand, benzene has six carbons and six p-electrons accessible for metal coordination and bond formation. The vast majority of metal/arene complexes exhibit h6- coordination with equal metal–carbon distances and equal contributions of all p-orbitals to the coordination bonding [2]. In contrast, arene ligands of lower hapticity exhibit aromatic p- orbitals that are no longer equivalent. As a consequence, the orbital symmetry of the benzene ring is distorted up to the point of complete loss of aromaticity, and the arene ligands become quite reactive. As such, h1- and h2-complexes represent the most interesting substrates for the study of metal–ligand bonding, exhibiting the most pronounced chargetransfer e ects, although such reactive complexes are di cult to isolate for X-ray crystallographic analysis [2].
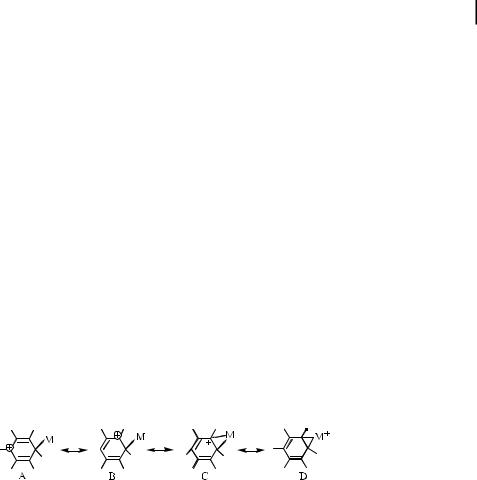
In spite of the large di erence between the h1- and h2-complexes from a structural point of view, their electronic configurations are closely related since two electrons are involved in the coordination bond in both types of complexes. The resonance structure C betrays the facile interconversion between these coordination types [2], i.e.:
Scheme 1
h1-Coordination leads to polarization of the p-electrons of the aromatic ligand, whereas h2- coordination does not necessarily lead to a polar electronic configuration. Because polarized structures are expected to predominate in the complexes containing metal and arene ligands of very di erent donor/acceptor strengths, the coordination type depends strongly on the relative donor/acceptor strengths of the arene ligand and the metal center. The transition from h2- to h1-coordination with increasing acceptor strength is observed in the series of arene complexes with di erent organic, inorganic, and organometallic acceptors. For example, the molecular complex between durene and CBr4 as the s-acceptor features a bridging bromine between neighboring ring carbons of the durene, with carbon–bromine distances of 3.26 and 3.34 A˚ . Moreover, in most AgI complexes with arenes, the AgI lies in a bridging position between two carbon atoms with equal CaAgI distances of about 2.5 A˚ . Moderately stronger acceptors such as CuI, Ni0, HgII, etc. still prefer h2-coordination. For example, in m-h2 Ni0/BEN, Cuþ/BEN, and Hg2þ/HMB complexes, the distances between the bridging

44813 Charge-Transfer Effects on Arene Structure and Reactivity
metals and the closest carbon atom in the aromatic ring are 2.005 and 2.021 A˚ (to Ni0), 2.081 and 2.105 A˚ (to CuI), and 2.56 and 2.58 A˚ (to HgII). In contrast, strong electron acceptors such as Al(C6F5)3, SiEt3þ, Brþ, and Clþ form exclusively h1-coordinated arene complexes. Similarly, strong organometallic acceptors such as perfluoroaryl-coordinated PdII or PtII
form mostly h1-complexes, while, in similar complexes, the weaker Mo(CO)3 acceptor shows a clear indication of h2-coordination with MoaC distances of 2.776(4) and 2.840(4) A˚ for ipso-
and ortho-coordination, respectively. A similar dependence of coordination type on the relative donor/acceptor strengths is observed for a variety of complexes. For example, bivalent d- metals from CrII to NiII show h1-coordination with the mesitylene group, while ReI, RhI, IrI, and Ni0 form h2-coordinated complexes with the electron-acceptor hexafluorobenzene acting as Lewis base. In many cases, h1- and h2-coordinated isomers can be very close in energy, and thus exhibit fast rates of h1-h2 interconversion in solution, which can be shifted by changing the relative donor–acceptor strengths of the complex. Dual arene reactivity toward both electrophiles as well as nucleophiles is possible. Thus, the more polarized h1-isomer with a positive charge residing on the benzene ring shows enhanced reactivity toward nucleophiles, while the h2-isomer exhibits high electron density at localized double bonds and is thus susceptible to electrophilic attack [2].
Scheme 2
13.3.3
E ect of Charge Transfer on the Structural Features of Coordinated Arenes
13.3.3.1 Expansion of the Arene Ring
In the framework of MO theory, both electron detachment from the benzene (p-bonding) HOMO and the addition of an electron to the (p -antibonding) benzene LUMO will lead to a decrease in the average bond order in the arene due to loss (or annihilation) of one bonding electron. Consistent with Pauling’s direct relationship between bond order and bond length [35], expansion of the arene ring is observed both when the arene acts as an electron donor (and su ers from electron deficiency) or is an electron acceptor (with partial population of the antibonding orbital). However, the changes in the benzene bond lengths upon one-electron oxidation or reduction are calculated to be only about 0.01 A˚ due to the involvement of six CaC bonds with twelve p-bonding electrons [2]. Therefore, the vast majority of organometallic structures are not suitable models for detecting electron deficiencies or partial cation-radical character in an arene ligand. However, recent advances in the use of lowtemperature X-ray di ractometers (with up to 0.001 A˚ precision) have enabled neutral arenes and their cation (anion) radicals to be distinguished on the basis of di erences in their average CaC bond distances. Highly precise X-ray structures clearly reveal significant ex-


450 13 Charge-Transfer Effects on Arene Structure and Reactivity
However, h2-coordination alone does not result in any degradation of the delocalized electronic configuration, and the above model involves bond localization only in the case of a significant degree of charge transfer between the aromatic ligand and the metal center. For example, in very weak [DUR, CBr4] and [BEN, Agþ]ClO4 complexes, very low degrees of charge transfer occur, and only subtle changes in the geometry (i.e. double-bond localization, and sp2/sp3 rehybridization) of the arene can be detected. Additionally, a bridging arene positioned between two acceptors is frequently observed, which leads to m-h2 (1,2):(4,5) complexes. In these complexes, the two metal centers induce opposite changes in the geometry of the benzene ring, which annihilate each other. Stronger metal acceptors (e.g. Mo0 and PtII) more clearly promote the formation of localized double bonds in the h2-coordinated benzene ring. Bond contraction of two non-coordinated p-bonds to lengths of just 1.37 A˚ and 1.35 A˚ was found in substituted benzene ligands coordinated to Mo0 and PtII, respectively. These distances correspond to bond orders of 1.7 and 1.9. When stronger arene donors, such as naphthalene or anthracene, are coordinated to RuI or Ni0 centers, the non-coordinated C(a)aC(b) bond clearly approaches the standard value of 1.34 A˚ for a normal CbC double bond [2].
Localized double bonds, in turn, represent attractive targets for further attack by a second electrophilic center, which explains the frequent observation of m-h2(1,2):h2(3,4) complexes. Such (1,2):(3,4) coordination results in cumulative e ects on the geometry of the benzene ring and the strength of the metal–carbon bond (as opposed to an annihilative e ect in the (1,2):(4,5) compounds mentioned above). For example, m-h2(1,2):h2(3,4) coordination in the Ni0 complex leads to MaC bonds that are close in length to a single s-bond [2]. It should be noted that such an e ect involving the relocation of roughly 0.5 electrons within the aromatic ring is connected with the acceptor strength of nickel, since the bond-length distribution in the isoelectronic copper complex is quite di erent [2].
Scheme 4
In most h1- and h2-coordinated complexes, the arene ligand acts as the Lewis base and electron donor, whereas the metal center acts as the Lewis acid and electron acceptor. Thus, a (formal) two-electron donation and (actual) charge transfer occurs in the same direction from the arene to the metal. However, when the acceptor strength of the arene is higher than that of the metal, coordination and charge transfer occur in opposite directions. For example, hexafluorobenzene donates two electrons in coordinating to a metal center. However, its complexes with metals having medium donor/acceptor properties (e.g. RhI, Ni0, ReI, IrI) exhibit substantial (back-) charge transfer from the metal to the arene that strengthens the bonding in these sðh2Þ-complexes. As the result, the arene ligand shows a clear cyclohexadienyl structure, the fluorine atoms at the ipso carbons deviate from the plane of the ring by as much as 48 , and the metal–carbon bonds are even shorter than those of


45213 Charge-Transfer Effects on Arene Structure and Reactivity
the plane of the aromatic ring (due to a transition from sp2 to sp3 hybridization); (iii) redistribution of the bond lengths in the benzene ring, i.e. elongation of the 1,2-bond to the standard distance of 1.50 A˚ for a C(sp2)aC(sp3) single bond and partial localization of the 2,3-double bond, and (iv) an increase in the degree of charge transfer, as in the chloronium and bromonium complexes with hexamethylbenzene, in which the positive charge of the inorganic component is completely transferred to the arene.
Similar e ects are observed in h2-complexes; AgI and HgII coordinated to arenes exhibit bending angles of a A5 (which is symptomatic of high p-character of the complex and a low degree of CT). On the other hand, in rhenium, rhodium, iridium, and nickel complexes, arenes exhibit bending angles of up to 45 [2].
In its extreme case, the CT interaction in arene/metal complexes also a ects the planarity of the arene ring itself. Various degrees of ring folding are observed in h4-coordinated complexes of benzene with metal centers, ranging from folding angles of b ¼ 0 in alkaline metal salts, to b ¼ 15 in lanthanide complexes, and to b ¼ 30–35 in the early transition metal complexes (e.g. with titanium, zirconium). The highest degree of folding is found in the iron and ruthenium complexes ðb ¼ 40 Þ. Interestingly, arene complexes with late transition metals such as cobalt ðb ¼ 35 Þ and nickel (b ¼ 20–25 ) show lower folding angles [2].
Scheme 5
In most h6-coordinated d-metal complexes, the six-carbon ring of benzene remains more or less planar, and its distortion is only manifested in the various degrees of bending of the substituents out of the ring plane. However, significant folding of the arene ring to a boat shape is observed in complexes of benzene with tantalum. Interestingly, the degree of folding depends on the oxidation state of the metal: 20 for TaII and 25 for TaIII complexes [2]. The better acceptor, TaIII, induces a stronger folding due to a greater degree of charge transfer from the benzene to the tantalum center, and consequently a higher degree of s- character in the arene–metal bond.
13.4
Charge-Transfer Activation of Coordinated Arenes
Redistribution of electron density in CT complexes results in a modification of the chemical properties of coordinated arenes, and this e ect is widely used in organometallic catalysis [2]. To demonstrate the relationship between charge transfer in arene complexes and their reactivity, we focus our attention on carbon–hydrogen bond activation, nucleophilic/ electrophilic umpolung, and the donor/acceptor properties of arenes in a wide variety of organometallic reactions.