

Astruc D. - Modern arene chemistry (2002)(en)
.pdf11.3 Cine and Tele Nucleophilic Aromatic Substitutions 393
Tab. 5. Cine and tele nucleophilic aromatic substitution of arenetricarbonylchromium complexes.
Entry |
Arene |
Nucleophile |
Ref., Year |
|
|
|
|
1 |
Diphenyl ether |
LiCMe2CN, LiCMe2CO2R |
75, 1985–86/89 |
2 |
p-MeC6H4Cl |
LiCMe2CN |
69a,e, 1986–89 |
3 |
2,6-diMe-C6H3X; X ¼ F, Cl |
LiC(CH3)[S(CH2)3S], LiCMe2CN |
72, 1987–89 |
|
|
LiC(Ph)[S(CH2)3S] |
|
4 |
m-MeC6H4Cl |
LiCMe2CN, LiC(Ph)[S(CH2)3S] |
69e, 1989 |
5 |
o-MeC6H4Cl |
ArSO2CH2Li |
71, 1990–94 |
6 |
o-Me3SiC6H4X |
LiEt3BD |
69f, 1991 |
7 |
o-Me3SiC6H4OPh |
LiEt3BD |
69f, 1991 |
8 |
o-Me3SiC6H4NMe2 |
LiEt3BD |
73, 1991 |
9 |
methoxyarene |
nBuLi |
58, 1997 |
10 |
veratrole |
LiCR2CN |
58, 77, 1997, 1989 |
11 |
1,3,5-trimethoxybenzene |
LiCR2CN |
74, 2000 |
|
|
|
|
For cine SNAr, see refs. [16, 69a, 71a, 71b, 73, 75b, 77].
For tele-meta SNAr, see refs. [16, 57, 58, 69b–d, 70, 73, 74, 75a, 75b, 76].
For tele-para SNAr, see: [72a, 72b].
1-methyl-4-chloro-5-(20-phenyl-10,30-dithiane)-1,3-cyclohexadiene 94b as the sole cyclohexadiene isomer, which has been characterized by X-ray crystallography [69b]. This study not only shows that the nucleophile adds ortho to the chloro substituent, but also that isomerization of the h4-cyclohexadiene 94 coordinated to Cr(CO)3 does not occur for steric reasons, thus avoiding elimination of HX.
Scheme 41. Cine and tele-meta SNAr: CaCl bond cleavage.
Addition of ArSO2CH2Li to o-chlorobenzenetricarbonylchromium complex 14a gives, after CF3CO2H treatment, the meta-disubstituted complex 95a in 72 % yield at 0 C under thermodynamic control (cine SNAr). Addition of the nucleophile, even at 10 C, occurs initially at the carbons C3 and C6 (Scheme 42) [71].
Addition of two equivalents of LiCMe2CN or LiC(Ph)[S(CH2)3S] to m-chlorotoluene- Cr(CO)3 complex 14b, followed by acid treatment, gives the tele-meta complexes 95b and 95c in yields of 62 % and 58 %, respectively. No incorporation of deuterium is observed, in good agreement with the mechanism described above (Scheme 5). This meta regioselectivity can

394 11 Arenetricarbonylchromium Complexes: Ipso, Cine, Tele Nucleophilic Aromatic Substitutions
Scheme 42. Cine and tele-meta SNAr: CaCl bond cleavage.
be explained in terms of preferential addition of the stabilized carbanion to the carbon eclipsed by a CraCO bond in the most stable conformer [69e]. This tele-meta regioselectivity is also observed on adding LiEt3BD to o-chloro- or o-fluorotrialkylsilylbenzenetricarbonyl- chromium complexes having an anti-eclipsed conformation with respect to the SiR3 group, giving p-deuterated trialkylsilylbenzenetricarbonylchromium complexes [69d].
The e ect of the nature of the leaving group has been delineated in the case of 2,6- dimethylfluoro and chloro complexes. Complex 96a reacts with LiC(CH3)[S(CH2)3S] and CF3CO2D to give exclusively complex 97a by a tele-meta SNAr [70], whereas complex 96b reacts with LiC(Ph)[S(CH2)3S] and CF3CO2D to give deuterated complex 97b by a tele-para SNAr [72].
Scheme 43. Tele-meta and tele-para SNAr: CaCl bond cleavage.
11.3.2
Cleavage of CxO Bonds
Reaction of NuLi (Nu ¼ CMe2CN, CHMeCN, CH2CN, etc.) with 5-deuterio-2,3-dimethyldi- phenylethertricarbonylchromium complex 98 yields, after CF3CO2H(D) treatment, the deuterated complex 99. This reaction has been shown to involve a 1,5-deuteride migration from the C5 carbon to the C1 carbon (Scheme 44) [75a, 75b].
Scheme 44. Tele-meta SNAr: CaO bond cleavage.

11.3 Cine and Tele Nucleophilic Aromatic Substitutions 395
Reaction of LiEt3BD and CF3CO2H with o-trimethylsilyldiphenylethertricarbonylchro- mium complex 100 gives the tele-meta complex 101 in 77 % yield (Scheme 45) [69d].
Scheme 45. Tele-meta SNAr: CaO bond cleavage.
Treatment of veratroletricarbonylchromium complex 102 with LiCH2CN in THF at 40 C for 30 min and then with acid leads to the formation of (3-methoxyphenyl)acetonitrile complex 103a in 47 % yield (Scheme 46). From this experiment, it cannot be ascertained whether the carbanion adds to the carbon ortho to the methoxy group (cine SNAr) or to the carbon para to the methoxy group (tele-para SNAr) because in each case a meta-disubstituted complex is formed. Consequently, CF3CO2D is used, whereupon complex 103b deuterated at the C2 carbon is obtained, as befits a cine SNAr. When the positions ortho to the MeO groups are substituted, tele regioselectivity is observed [76]. The same reaction with 1,2,3-trimethoxy- benzenetricarbonylchromium complex 71 a ords complex 72 by a tele-meta SNAr as the major product (42 % yield), contaminated by the ipso complex 73 (22 % yield) (vide supra; Scheme 34) [58, 74].
Scheme 46. Cine SNAr: CaO bond cleavage.
11.3.3
Cleavage of CxN Bonds
An unprecedented cleavage of a carbon–nitrogen bond of a tertiary amine is observed on treating o-trimethylsilyldimethylanilinetricarbonylchromium complex 104a with LiEt3BD and then with CF3CO2H. Complexes 105a and 105b (ratio 20:80) are recovered as a result of tele-meta SNAr (20:80 ratio; 55 % yield; Scheme 47) [73]. When the same experiment is
Scheme 47. Cine and tele SNAr: CaN bond cleavage.

39611 Arenetricarbonylchromium Complexes: Ipso, Cine, Tele Nucleophilic Aromatic Substitutions
carried out with the meta isomer 104b, complex 105a is formed, which can be explained in terms of a cine mechanism after incorporation of a second deuterium at the meta position relative to the SiMe3 group if CF3CO2D is used.
11.4
Concluding Remarks
Methods are available using arenetricarbonylchromium complexes that allow the substitution of hydrogen, halogen, alkoxy, aryloxy, and amino groups by nucleophiles under very mild conditions without decoordination of the Cr(CO)3 unit. In the light of the results presented herein, three new SNAr processes, namely the cine, tele-meta, and tele-para SNAr, have been discovered in our laboratory, which broaden the scope of the applications of these complexes in organic chemistry. These reactions are possible by treating an arenetricarbonylchromium complex substituted by a good leaving group first with a nucleophile and then with an acid. This represents an alternative to the well-known ipso SNAr, which corresponds to a direct addition of a nucleophile to the carbon bearing a leaving group. Knowing that Cr(CO)6 can be recovered by treating these (h6-arene) complexes under CO atmosphere, it is possible to recover the free arene as well as Cr(CO)6 necessary to prepare the chromium complex. This is the first step of a ‘‘pseudo catalytic’’ cycle. Further work will focus on the possibility of transforming these stoichiometric reactions into catalytic reactions. The main challenge at the present time is to find a general process whereby these fantastic creations of CaO, CaN, and CaC bonds can be made catalytic in the metal. It is also fascinating to know that it is now possible to predict the regioselectivity of the addition of a nucleophile, provided that the reaction is under kinetic control at low temperature. This sheds light on the fundamental role of the Cr(CO)3 conformation in solution, because addition of the nucleophile occurs preferentially at a carbon eclipsed by a CraCO bond. Knowing that the Friedel–Crafts reaction is widely used in arene chemistry to add an electrophile to a ring, it is clear that addition of a nucleophile represents a complementary tool for the functionalization of an arene ring, for which the regioselectivity is now predictable. Ipso, cine, tele-meta, and tele-para SNAr are reactions that keep the Cr(CO)3 entity still coordinated to the arene ring, and thus a second nucleophile can react to give new disubstituted cycles.
Abbreviations
Cp |
cyclopentadienyl |
DMSO |
dimethyl sulfoxide |
HMPA |
hexamethylphosphoramide |
LDA |
lithium diisopropylamide |
SNAr |
nucleophilic aromatic substitution |
TBAB |
tetrabutylammonium bromide |
THF |
tetrahydrofuran |
TMEDA |
tetramethylethylenediamine |

References 397
Acknowledgements
For the published and unpublished work from our group, we gratefully acknowledge our collaborators whose names appear in the cited references. We thank the CNRS and the EU training and mobility of researchers programme, contract ERBFMR XCT-CT98-0166, for financial support.
References
1 |
For reviews, see: a) L. Balas, D. Jhurry, |
7 |
a) S. I. Rosca, S. Rosca, Revista de Chemie |
|
L. Latxague, S. Grelier, Y. Morel, M. |
|
(Bucharest) 1964, 25, 461; b) M. Uemura, |
|
Hamdani, D. Astruc, Bull. Soc. Chim. Fr. |
|
H. Nishimua, T. Hayashi, J. Organomet. |
|
1990, 127, 401; b) F. J. McQuillin, D. G. |
|
Chem. 1994, 473, 129. |
|
N. Parker, G. R. Stephenson, Transition |
8 |
T. Oishi, M. Fukui, Y. Endo, Heterocycles |
|
Metals in Organic Synthesis, Cambridge |
|
1977, 7, 947. |
|
University Press, Cambridge, 1991; c) |
9 |
A. Alemagna, C. Baldoli, P. Del |
|
M. F. Semmelhack, in: Comprehensive |
|
Buttero, E. Licandro, S. Maiorana, |
|
Organometallic Chemistry II, vol. 12 (Eds.: |
|
Gazz. Chim. Ital. 1985, 115, 555. |
|
E. W. Abel, F. G. A. Stone, G. Wilkin- |
10 |
J. Hamilton, C. A. L. Mahaffy, Synth. |
|
son), Pergamon, Oxford, 1995, p. 979; |
|
React. Inorg. Met.-Org. Chem. 1986, 116, |
|
d) S. G. Davies, T. D. McCarthy, in |
|
66. |
|
Comprehensive Organometallic Chemistry II, |
11 |
F. Rose-Munch, R. Khourzom, J.-P. |
|
vol. 12 (Ed.: E. W. Abel, F. G. A. Stone, |
|
Djukic, E. Rose, J. Organomet. Chem. |
|
G. Wilkinson), Pergamon, Oxford, 1995, |
|
1993, 456, C8. |
|
p. 1039; e) M. J. Morris, in Comprehensive |
12 |
A. Alemagna, C. Baldoli, P. Del |
|
Organometallic Chemistry II, Vol. 5 (Eds.: |
|
Buttero, E. Licandro, S. Maiorana, J. |
|
E. W. Abel, F. G. A. Stone, G. Wilkin- |
|
Chem. Soc., Chem. Commun. 1985, 417. |
|
son; Volume Eds.: J. A. Labinger, M. J. |
13 |
A. Alemagna, C. Baldoli, P. Del |
|
Winter), Pergamon, Oxford, 1995; f ) F. |
|
Buttero, E. Licandro, S. Maiorana, |
|
Rose-Munch, V. Gagliardini, C. |
|
Synthesis 1987, 192. |
|
Renard, E. Rose, Coord. Chem. Rev. 1998, |
14 |
C. Baldoli, P. Del Buttero, E. |
|
178, 249; g) F. Rose-Munch, E. Rose, |
|
Licandro, S. Maiorana, Synthesis 1988, |
|
Curr. Org. Chem. 1999, 3, 445; h) F. Rose- |
|
344. |
|
Munch, E. Rose, J. P. Djukic, J. Vaisser- |
15 |
D. G. Loughhead, L. A. Flippin, R. J. |
|
mann, Eur. J. Inorg. Chem. 2000, 1295; |
|
Weikert, J. Org. Chem. 1999, 64, 3373. |
|
i) A. R. Page, K. P. Kaliappan, E. P. |
16 |
F.-E. Hong, S.-C. Lo, M.-W. Liou, Y.-T. |
|
Ku¨ndig, Chem. Rev. 2000, 100, 2917; j) D. |
|
Chang, C.-C. Lin, J. Organomet. Chem. |
|
Astruc, in Chimie Organome´tallique, EDP |
|
1996, 506, 101 |
|
Sciences, Les Ulis, France, 2000; Chapters |
17 |
F. Hossner, M. Voyle, J. Organomet. |
|
3–6, p. 87; k) F. Rose-Munch, E. Rose, |
|
Chem. 1988, 347, 365. |
|
Eur. J. Inorg. Chem. 2002, 1269. |
18 |
a) R. P. Houghton, M. Voyle, J. Chem. |
2 |
D. Astruc, Top. Curr. Chem. 1992, 160, 47. |
|
Soc., Chem. Commun. 1980, 884; b) R. P. |
3 |
L. A. P. Kane-Maguire, E. D. Honig, |
|
Houghton, M. Voyle, R. Price, J. |
|
D. A. Sweigart, Chem. Rev. 1984, 84, |
|
Organomet. Chem. 1983, 259, 183. |
|
525. |
19 |
C. Baldoli, P. Del Buttero, S. |
4 |
R. D. Pike, D. A. Sweigart, Synlett. 1990, |
|
Maiorana, A. Papagni, J. Chem. Soc., |
|
565. |
|
Chem. Commun. 1985, 1181. |
5 |
C. A. L. Mahaffy, P. L. Pauson, Inorg. |
20 |
M. F. Semmelhack, J. Bisaha, M. |
|
Synth. 1979, 19, 154. |
|
Czarny, J. Am. Chem. Soc. 1979, 101, 768. |
6 |
B. Nicholls, M. C. Whiting, J. Chem. |
21 |
P. J. Beswick, D. A. Widdowson, |
|
Soc. 1959, 551. |
|
Synthesis 1985, 492. |

398 11 Arenetricarbonylchromium Complexes: Ipso, Cine, Tele Nucleophilic Aromatic Substitutions
22a) A. Alemagna, P. Del Buttero, C. Gorini, D. Landini, E. Licandro, S. Maiorana, J. Org. Chem. 1983, 48, 605; b) A. Alemagna, P. Cremonesi, P. Del Buttero, E. Licandro, S. Maiorana, J. Org. Chem. 1983, 48, 3114.
23C. Baldoli, P. Del Buttero, E. Licandro, S. Maiorana, Gazz. Chim. Ital.
1988, 118, 409.
24M. J. Dickens, J. P. Gilday, T. J. Mowlem, D. A. Widdowson, Tetrahedron
1991, 47, 8621.
25A. A. Vasil’ev, L. Engman, J. P. Storm, C.-M. Anderson, Organometallics 1999, 18, 1318.
26a) A. N. Nesmeyanov, N. A. Vol’kenau, I. N. Boleseva, Dokl. Akad. Nauk. SSSR
1967, 175, 1606 and 1968, 183, 834; b)
J. F. Bunnet, H. Hermann, J. Org. Chem.
1971, 36, 4081.
27C. A. L. Mahaffy, P. L. Pauson, J. Chem. Res. 1979, 128.
28a) M. Ghavshou, D. A. Widdowson, J. Chem. Soc., Perkin Trans. 1 1983, 3065; b) J. P. Gilday, D. A. Widdowson, J. Chem. Soc., Chem. Commun. 1986, 1235.
29J. P. Gilday, D. A. Widdowson,
Tetrahedron Lett. 1986, 27, 5525.
30Y. Ishii, Y. Ishino, T. Aaoki, M. Hidai, J. Am. Chem. Soc. 1992, 114, 5429.
31C. Baldoli, P. Del Buttero, S. Maiorana, Tetrahedron. Lett. 1992, 33, 4049.
32S. G. Davies, W. E. Hume, Tetrahedron Lett. 1995, 36, 2673.
33J. P. Djukic, F. Rose-Munch, E. Rose, Y. Dromzee, J. Am. Chem. Soc. 1993, 115, 6434.
34M. Perez, P. Potier, S. Halazy,
Tetrahedron Lett. 1996, 37, 8487.
35M. F. Semmelhack, G. Hilt, J. H. Colley, Tetrahedron Lett. 1998, 39, 7683; see also refs. [27, 29].
36K. G. Molay, J. L. Petersen, J. Am. Chem. Soc. 1995, 117, 7696.
37S. Maiorana, C. Baldoli, P. Del Buttero, M. Di Ciolo, A. Papagni,
Synthesis 1998, 5, 735.
38S. Maiorana, C. Baldoli, E. Licandro, L. Casiraghi, E. de Magistris, A. Paio, S. Provera, P. Serreci, Tetrahedron Lett.
2000, 41, 7271.
39M. R. G. Da Costa, M. J. M. Curto, S. G. Davies, M. T. Duarte, C. Resende, F. C.
Teixeira, J. Organomet. Chem. 2000, 604, 157.
40a) G. R. Brown, A. J. Foubister, P. D. Ratcliffe, Tetrahedron Lett. 1999, 40, 1219; b) A. J. Belfield, G. R. Brown,
A.J. Foubister, P. D. Ratcliffe, Tetrahedron 1999, 50, 1328; c) M. Kidwai,
P.Sapra, B. Dave, Synth. Commun. 2000,
30, 4479.
41Pd: a) A. S. Guram, S. L. Buchwald, J. Am. Chem. Soc. 1994, 116, 7901; b) F. Paul, J. Patt, J. F. Hartwig, J. Am. Chem. Soc. 1994, 116, 5969; c) L. M. AlcazarRoman, J. F. Hartwig, A. L. Rheingold,
L.M. Liable-Sands, I. A. Guzei, J. Amer. Chem. Soc. 2000, 122, 4618; d) F. A. Hicks, M. Brookhart, Org. Lett. 2000, 219; e) J. P. Wolfe, S. L. Buchwald, J.
Org. Chem. 2000, 65, 1144; f ) S. R. Stauffer, S. Lee, J. P. Stambuli, S. I. Hauck, J. F. Hartwig, Org. Lett. 2000, 2; 1423, Ni: g) C. Desmarets, R. Schneider, Y. Fort, Tetrahedron Lett. 2001, 42, 247; h) E. Brenner, R. Schneider, Y. Fort,
Tetrahedron Lett. 2000, 41, 2881; i) A. J. Belfield, Tetrahedron 1999, 50, 1328; Cu: j) R. K. Gujadhur, C. G. Bates, D. Venkataraman, Org. Lett. 2001, 3, 4315; without metal activation, see ref. [40].
42S. G. Nelson, M. A. Hilfiker, Org. Lett.
1999, 1, 1379.
43D. Brown, J. R. Raju, J. Chem. Soc., A
1966, 40 and 1617.
44I. U. Khand, P. L. Pauson, W. E. Watts,
J.Chem. Soc., C 1968, 2257.
45a) M. F. Semmelhack, H. T. Hall, J. Am. Chem. Soc. 1974, 96, 7091; b) M. F. Semmelhack, H. T. Hall, J. Am. Chem. Soc. 1974, 96, 7092; c) M. F. Semmelhack,
H.T. Hall, M. Yoshifuji, G. Clark, J. Am. Chem. Soc. 1975, 97, 1247.
46J. C. Boutonnet, F. Rose-Munch, E. Rose, A. Semra, Bull. Soc. Chim. Fr. 1987, 640.
47H. Ku¨nzer, M. Thiel, Tetrahedron Lett.
1988, 29, 1135.
48A. Ceccon, A. Gambaro, F. Gottardi, F. Manoli, A. Venzo, J. Organomet. Chem.
1989, 363, 91.
49M. Chaari, J.-P. Lavergne, P. Viallefont, Synth. Commun. 1989, 19, 1211.
50M. Chaari, A. Jenki, J.-P. Lavergne, Ph. Viallefont, J. Organomet. Chem. 1991,
401, C10.
|
|
|
References |
399 |
|
F. Rose-Munch, K. Aniss, E. Rose, J. |
|
|
|
51 |
68 |
J. Li, A. D. Hunter, R. McDonald, B. D. |
||
|
Organomet. Chem. 1990, 385, C1. |
|
Santarsiero, S. G. Bott, J. L. Atwood, |
|
52 |
F. Rose-Munch, K. Aniss, E. Rose, J. |
|
Organometallics 1992, 11, 3050. |
|
|
Vaissermann, J. Organomet. Chem. 1991, |
69 |
a) F. Rose-Munch, E. Rose, A. Semra, J. |
|
|
415, 223. |
|
Chem. Soc., Chem. Commun. 1986, 1551; b) |
|
53 |
J.-C. Boutonnet, O. Le Martret, L. |
|
F. Rose-Munch, E. Rose, A. Semra, Y. |
|
|
Mordenti, G. Precigoux, E. Rose, J. |
|
Jeannin, F. Robert, J. Organomet. Chem. |
|
|
Organomet. Chem. 1981, 221, 47. |
|
1988, 353, 53; c) F. Rose-Munch, E. Rose, |
|
54 |
F. Rose-Munch, L. Mignon, J.-P. |
|
A. Semra, C. Bois, J. Organomet. Chem. |
|
|
Souchez, Tetrahedron Lett. 1991, 32, 6323. |
|
1989, 363, 103; d) J.-P. Djukic, P. |
|
55 |
T. J. Henly, C. B. Knobler, M. F. |
|
Geysermans, F. Rose-Munch, E. Rose, |
|
|
Hawthorne, Organometallics 1992, 11, |
|
Tetrahedron Lett. 1991, 32, 6703. |
|
|
2313. |
70 |
F. Rose-Munch, E. Rose, A. Semra, L. |
|
56 |
a) K. Kamikawa, M. Uemura, Tetrahedron |
|
Mignon, J. Garcia-Oricain, C. Knobler, |
|
|
Lett. 1996, 37, 6359; b) K. Kamikawa, M. |
|
J. Organomet. Chem. 1989, 363, 297. |
|
|
Uemura, Synlett 2000, 7, 938. |
71 |
a) F. Rose-Munch, J.-P. Djukic, E. Rose, |
|
57 |
F.-E. Hong, Y.-C. Yong, S.-C. Lo, C.-C. |
|
Tetrahedron Lett. 1990, 31, 2011; b) F. |
|
|
Lin, Polyhedron 1997, 16, 2005. |
|
Rose-Munch, R. Khourzom, J.-P. |
|
58 |
V. Gagliardini, V. Onnikian, F. Rose- |
|
Djukic, A. Perrotey, E. Rose, J. |
|
|
Munch, E. Rose, Inorg. Chim. Acta 1997, |
|
Brocard, J. Organomet. Chem. 1994, 467, |
|
|
259, 265. |
|
195. |
|
59 |
T. Mino, T. Matsuda, K. Maruhashi, M. |
72 |
a) F. Rose-Munch, E. Rose, A. Semra, M. |
|
|
Yamashita, Organometallics 1997, 16, |
|
Philoche-Levisalles, J. Organomet. Chem. |
|
|
3241. |
|
1989, 363, 123; b) F. Rose-Munch, E. |
|
60 |
S. Varela Calafat, E. N. Durantini, J. J. |
|
Rose, A. Semra, J. Chem. Soc., Chem. |
|
|
Silber, S. M. Chiacchiera, |
|
Commun. 1987, 942. |
|
|
Organometallics 1999, 18, 2727. |
73 |
J.-P. Djukic, F. Rose-Munch, E. Rose, J. |
|
61 |
F. Rose-Munch, J.-P. Djukic, E. Rose, |
|
Chem. Soc., Chem. Commun. 1991, 22, |
|
|
Tetrahedron Lett. 1990, 31, 2589. |
|
1634. |
|
62 |
M. Persson, U. Hacksell, I. Csoregh, J. |
74 |
F. Rose-Munch, R. Chavignon, J.-P. |
|
|
Chem. Soc., Perkin Trans. 1 1991, 1453. |
|
Tranchier, V. Gagliardini, E. Rose, |
|
63 |
J.-P. Djukic, F. Rose-Munch, E. Rose, |
|
Inorg. Chim. Acta 2000, 300–302, 693. |
|
|
Organometallics 1995, 14, 2027. |
75 |
a) J.-C. Boutonnet, F. Rose-Munch, E. |
|
64 |
J. A. Heppert, M. E. Thomas-Miller, |
|
Rose, Tetrahedron Lett. 1985, 26, 3989, b) |
|
|
P. E. Sweston, M. W. Extine, J. Chem. |
|
F. Rose-Munch, E. Rose, A. Semra, J. |
|
|
Soc., Chem. Commun. 1988, 280. |
|
Chem. Soc., Chem. Commun 1986, 1108; |
|
65 |
J. A. Heppert, M. A. Morgenstern, |
|
see also refs. [16, 76, 77]. |
|
|
D. M. Scherubel, F. Takusagawa, |
76 |
a) H.-G. Schmalz, M. Arnold, J. |
|
|
M. R. Shaker, Organometallics 1988, 7, |
|
Hollande, J. W. Bats, Angew. Chem. Int. |
|
|
1715. |
|
Ed. Engl. 1994, 33, 109; b) H.-G. Schmalz, |
|
66 |
J. A. Heppert, M. E. Thomas-Miller, |
|
K. Schellhaas, Tetrahedron Lett. 1995, 36, |
|
|
D. M. Scherubel, F. Takusagawa, M. A. |
|
5511; c) H.-G. Schmalz, K. Schellhaas, |
|
|
Morgenstern, M. R. Shaker, |
|
Angew. Chem. Int. Ed. Engl. 1996, 35, 2146; |
|
|
Organometallics 1989, 8, 1199. |
|
d) H.-G. Schmalz, T. Volk, D. Bernicke, |
|
67 |
G. B. Richter-Addo, A. D. Hunter, N. |
|
S. Huneck, Tetrahedron 1997, 53, 9219. |
|
|
Wichrowska, Can. J. Chem. 1990, 68, |
77 |
F. Rose-Munch, E. Rose, A. Semra, J. |
|
|
41. |
|
Organomet. Chem. 1989, 377, C9. |
|

400
12
Activation of Simple Arenes by the CpFeB Group and Applications to the Synthesis of Dendritic Molecular Batteries
Didier Astruc, Sylvain Nlate, and Jaime Ruiz
Abstract
The CpFeþ-induced polyfunctionalization of polymethylbenzenes under ambient conditions gives virtually quantitative yields of CpFeþ(h6-arene)-centered stars and dendritic cores on a large scale. From the chlorotoluene complex, mild nucleophilic reaction with ethanol in the presence of K2CO3 followed by mild reaction with allyl bromide in the presence of tBuOK gives a 60 % yield of the triallyl phenol dendron p-HOC6H4C(allyl)3. This dendron and its functionalized derivatives have been used as building blocks for the divergent synthesis of large dendrimers and for the convergent synthesis of nonafunctional dendrons including dendrons bearing ferrocenylsilyl termini on their branches. The various dendrimers, with branch numbers of up to 243 in theory, have been decorated with ferrocenyl groups, and polyaminodendrimers have been functionalized with ferrocenyl, cobaltocenyl, and CpFeþ- arene moieties. The generation or isolation of two redox states of these metallodendrimers and electron-transfer reactions illustrate their function as molecular electron reservoirs or molecular batteries.
12.1
Introduction
Molecular batteries should be useful as part of nanoscopic molecular electronic devices [1–4]. Ideally, nanoscopic assemblies containing a large number of redox centers, for which both redox forms are stable and which operate at the same redox potential, should be able to deliver or accept a large number of electrons at once at this redox potential. Such an assembly constitutes a battery – a molecular battery or a molecular electron reservoir if it is a molecular assembly. Metallodendrimers [5–14] are excellent candidates for such a function when at least two of their redox states are stable [15]. This property is realized with some late transition metal metallocenes and analogous sandwich systems. We describe herein the synthesis and properties of such nanoscopic metallo-assemblies that can be considered as molecular batteries. These syntheses are largely based on the activation of simple arenes by the electron-withdrawing group CpFeþ [16]. This metal group is thus useful for both
Modern Arene Chemistry. Edited by Didier Astruc
Copyright 8 2002 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim ISBN: 3-527-30489-4
12.2 General Features of the CpFeþ Activation of Arenes 401
dendritic construction and redox activity [5]. Below are summarized the main properties of the [CpFeþ(h6-arene)] complexes [17], with emphasis on those relevant to the dendritic syntheses and dendritic redox function.
12.2
General Features of the CpFeB Activation of Arenes
12.2.1
Complexation and Decomplexation
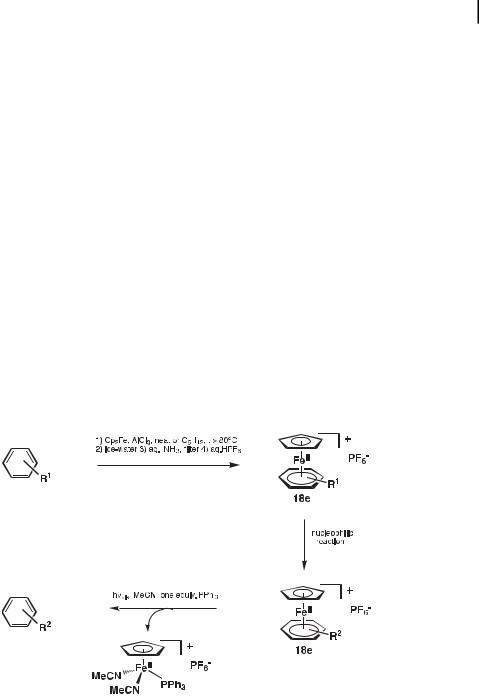
The CpFeþ group is an extremely powerful and useful activator of simple arenes [16–19]. It can be easily introduced on a large number of arenes either by reaction with ferrocene in the presence of aluminum chloride [20] or by photolytic arene exchange with another CpFeþ(h6- arene) complex in dichloromethane in the presence of visible light [18]. Following CpFeþ- activation of the desired reaction, the CpFeþ group can be easily removed by photolysis with visible light [21] in acetonitrile solution containing one equivalent of PPh3, and then the free arene can be extracted into pentane or diethyl ether (Scheme 1) [16]. Thus, although it is possible to work with organic solutions of these salts in daylight, prolonged exposure to light (especially sunlight) should be avoided if the purpose is not decomplexation. The CpFeþ(h6- aniline)-type complexes are resistant to decomplexation using this procedure, however, and decomplexation is best achieved by single-electron reduction to the unstable FeI complex using a rather strong reductant (with E more negative than 2 V vs. FeCp2) [22].
Scheme 1. Principle and general steps of the CpFeþ-induced activation of arenes by temporary complexation.
40212 Activation of Simple Arenes by the CpFeþ Group
12.2.2
Solubility, Stability, and General Reactivity Trends
The yellow CpFeþ(h6-arene) salts (most commonly BF4 or PF6 ) are usually stable up to at least 200 C, are stable in concentrated sulfuric acid, and are very resistant towards oxidation
(until recently, it was believed that they could not be oxidized [23]; vide infra). They are not easy to reduce either [23] (vide infra). The chloride salts [CpFeþ(h6-arene)] Cl are water-soluble; they are formed upon hydrolysis following ligand-exchange reactions between ferrocene and the arene in the presence of aluminum chloride [21]. Such aqueous solutions may sometimes
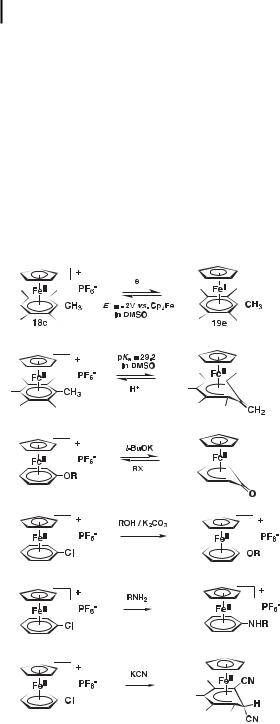
be directly used for nucleophilic reactions [22] (vide infra). The BF4 salts are also sometimes quite soluble in water, but the PF6 salts are much less so. Electrophilic reactions that are readily undergone by the free arenes, such as Friedel–Crafts reactions, are no longer possible on the CpFeþ(h6-arene) complexes [19, 23]. On the other hand, a range of nucleophilic re-
actions that are impossible or very di cult to carry out with free arenes become possible under ambient or mild conditions with the CpFeþ(h6-arene) complexes (Scheme 2) [16–20].
Scheme 2. Main reactions of the [CpFe(h6-arene)]þ complexes.