

Astruc D. - Modern arene chemistry (2002)(en)
.pdf8.3 Scope and Limitations 263
Scheme 15. Benzannulation of a fluorovinyl carbene complex.
8.3.1.3 The Chromium Template
The overwhelming majority of benzannulations with chromium carbene complexes involve compounds in which the metal bears only carbonyl ligands. However, phosphine ligands may also be present in the chromium coordination sphere.
The e ect of phosphine ligands di ering in their donor–acceptor properties on the benzannulation reaction has been studied with chromium carbenes (33a,b) (Scheme 16) [36a]. The yield of the tricarbonylchromium complex 2 starting from the pentacarbonyl carbene complex 1 (62 %) was essentially unchanged at a slightly higher reaction temperature (60 C for 33a compared to 45 C for 1) when a cis-CO ligand was replaced by tris(p-fluorophenyl)phosphine (33a). In contrast, the yield significantly dropped and the reaction temperature had to be increased to 90 C when the tris(n-butyl)phosphine complex 33b was subjected to the benzannulation reaction.
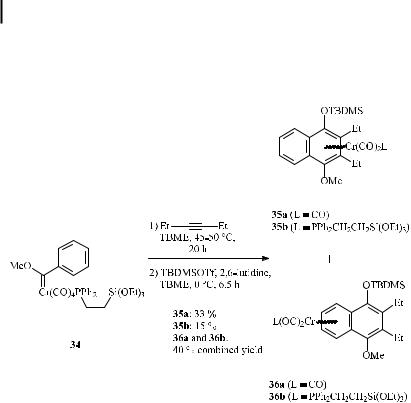
Very recently, the benzannulation has been applied to chromium(phosphine)carbonyl complex 34, which is suited for subsequent immobilization by sol-gel methodology; the tri-
Scheme 16. E ect of phosphine ligands on the benzannulation of methoxy phenyl carbene complexes with diphenylethyne.
2648 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
carbonylchromium complexes 35a (33 %) and 35b (15 %) were obtained, along with the dicarbonyl(phosphine)chromium complexes 36a and 36b (inseparable mixture in 40 % combined yield) (Scheme 17) [42].
Scheme 17. Synthesis of dicarbonyl(phosphine)carbene complexes by means of the benzannulation reaction.
Finally, the a,b-unsaturated carbene complex may be generated in situ by alkyne insertion into a chromium–carbene bond of a saturated chromium carbene leading to a chromium vinyl carbene (equivalent to intermediate (E )-D in the mechanism of the benzannulation reaction, see Section 8.2.1, Scheme 3), which may undergo subsequent benzannulation with a second equivalent of the alkyne [43a]. This strategy was subsequently applied to the synthesis of (Z )-enediynes and related compounds [43b], and to that of substituted benzofurans (see also Section 8.5) [43c, 43d].
8.3.2
The Alkyne
The benzannulation reaction tolerates a range of alkyl and aryl alkynes, which may bear additional functionalities. The simultaneous presence of two bulky substituents directly attached to the CcC bond, as for example in bis(trimethylsilyl)ethyne, however, blocks the final electrocyclization and causes the reaction to stop at the vinyl ketene stage [44]. Neither very electron-rich nor very electron-poor alkynes can undergo benzannulation. Strongly electron-deficient alkynes such as hexafluorobut-2-yne cannot adequately compete with car-

8.3 Scope and Limitations 265
bon monoxide in the initial ligand-exchange process. On the other hand, strongly electronrich alkynes, especially those having a polarized CcC bond such as ynamines, tend to preferentially add to the carbene carbon atom and subsequently insert into the metal–carbene bond. The insertion products may undergo cyclopentannulation at elevated temperatures [45].
Few examples are known in which acetylenic ethers have been incorporated into benzannulation products [46]. Propargyl ethers have been reported to a ord variable amounts of indenes and furans depending on the carbon chain [47]. Other heteroatoms that support the benzannulation reaction when adjacent to the CcC bond in the alkyne are boron (see Section 8.3.6), silicon, and tin (see Section 8.3.3). Haloalkynes give only inseparable product mixtures.
Furthermore, the successful [3þ2þ1] cycloaddition of alkynes bearing a cyclopropane ring and a carbene complex unit has been reported. These benzannulations result in the formation of bimetallic naphthohydroquinone chromium tricarbonyl complexes [48]. Additionally, (non-strained) cyclic alkynes are potent reaction partners in the cycloaddition of chromium carbene complexes [49].
The CcC bond is distinctly more reactive towards the metal carbene than the CbC bond [50]. This distinction allows a benzannulation approach to vitamins K and E (see Section 8.6.1). Bis(alkynes) have been shown to participate in the [3þ2þ1] cycloaddition with either one or both of their triple bonds (see Section 8.5).
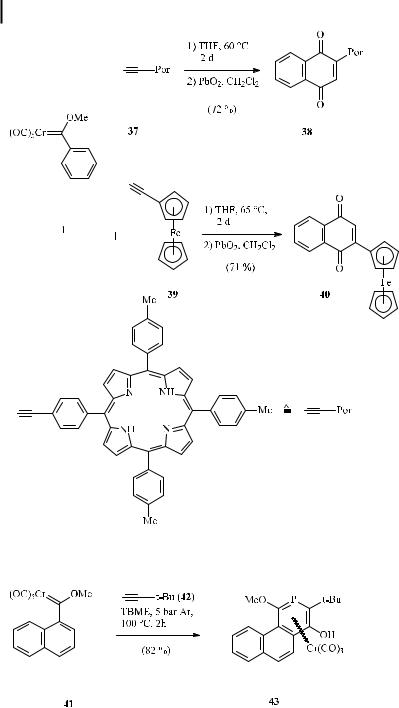
The benzannulation approach has been applied to aromatic molecules of biological interest. Alkynes bearing porphyrin (for example 37) [51a, 51b], ferrocene (for example 39) [51c], and carbohydrate functionalities [51d, 51e] have been incorporated into the (hydro)quinone skeleton, as demonstrated for porphyrinyl and ferrocenyl naphthoquinones 38 and 40 (Scheme 18).
tert-Butylphosphaethyne 42 is the only heteraalkyne known to be amenable to benzannulation [52]. In a competitive experiment, it was demonstrated [52c] that this phosphaalkyne is about six times more reactive than the corresponding tert-butylethyne. This is due to the higher electron density in tert-butylphosphaethyne.
A benzannulation was carried out with 42 and carbene complex 41 to give phosphaphenanthrene 43 in high yield (Scheme 19), whereas yields of phosphabenzenes were only marginal [52].
8.3.3
Regioselectivity
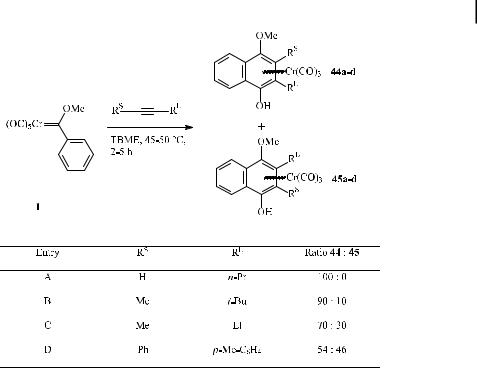
One of the main features of the benzannulation reaction of Fischer carbene complexes is the regiochemistry of the incorporation of alkynes into the assembled hydroquinone [53]. While terminal alkynes are incorporated with high regioselectivity (regardless of alkyl and aryl substituents), internal alkynes are prone to much poorer regioselectivity. Regioselectivity is virtually lost in the case of diarylacetylenes.
Scheme 20 presents the regiochemical outcome of some benzannulations with both terminal and internal alkynes. With similar alkyl substituents attached to the triple bond, the regioselectivity in favor of 44 gradually drops on going from 1-pentyne (entry A; exclusive formation of 44) through 2,2-dimethyl-3-pentyne (entry B; 44 : 45 ¼ 90 : 10) to 3-pentyne
266 8 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
Scheme 18. Applications of ferrocenyl and porphyrinyl alkynes in the benzannulation reaction.
Scheme 19. Synthesis of a phosphaphenanthrene via the benzannulation path.
8.3 Scope and Limitations 267
Scheme 20. Regioselective incorporation of terminal and internal alkynes in the benzannulation reaction.
(entry C; 44 : 45 ¼ 70 : 30), but incorporation of the larger alkyl substituent next to the phenolic hydroxy group remains favored. Entry D shows the poor selectivity observed when two similar aryl substituents are attached to the acetylene moiety; here, isomers 44 and 45 are obtained in comparable quantities.
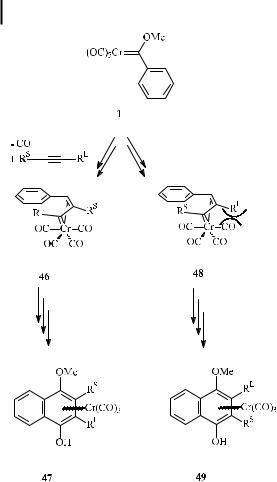
The regioselectivity is mainly governed by the steric demands of the two alkyne substituents (Scheme 21). The discrimination between the two possible regioisomers is supposed to occur at the stage of the vinyl carbene intermediates 46 and 48, respectively. While 48 su ers from steric repulsion between the apical carbonyl ligand and the larger RL substituent, this unfavorable interaction does not occur in regioisomer 46; as a result, regioisomer 47 generally prevails over 49.
The incorporation of alkynes bearing substituents of similar steric bulk next to the triple bond results in only poor regioselectivity. This restriction may be overcome by an intramolecular version of the benzannulation, in which the alkyne and the carbene complex are linked by a suitable spacer that may be detached upon oxidation [31, 54].
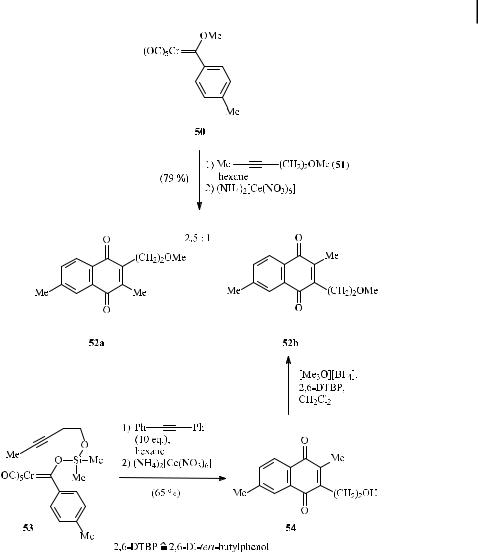
An advanced approach used a silyloxyethylene linker as in complex 53 (Scheme 22) [31]. Benzannulation in the presence of an excess of diphenylethyne (‘‘xenochemical e ect’’) gave naphthoquinone 54, which was subjected to oxidative cleavage and demetalation to a ord regioisomer 52b exclusively. The complementary intermolecular benzannulation protocol
268 8 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
Scheme 21. Origin of the regioselectivity of alkyne incorporation in benzannulation reactions.
involving chromium carbene 50 and alkyne 51 resulted in a 2.5 : 1 preference for the other naphthoquinone regioisomer 52a.
The regiochemistry of the intermolecular benzannulation can be reversed to some extent by an appropriate substitution pattern of the alkyne. While incorporation of silylacetylene occurs with the expected regiopreference in favor of the 2-silylphenols, the homologous stannylacetylene gives the reverse regiochemistry [55]. The origin of this reversal is not quite clear; it has been attributed to a stabilizing interaction between the tin and the adjacent apical carbon monoxide ligand at chromium in intermediate 48 (see Scheme 21) [55a]. A similar reversal of regiochemistry in the benzannulation product results from the ring-opening of arylcyclopropenes in the presence of hexacarbonylchromium [55b]. This rearrangement is believed to involve a metal-coordinated vinylcarbene intermediate, and thus parallels the benzannulation methodology of carbene complexes with alkynes.
8.3 Scope and Limitations 269
Scheme 22. Improved regioselectivity by intramolecular benzannulations.
8.3.4
Diastereoselectivity
The benzannulation a ords arene-Cr(CO)3 complexes possessing a plane of chirality resulting from the unsymmetrical arene substitution pattern. This aspect is relevant to stereoselective synthesis, in which enantiopure arene tricarbonyl chromium complexes play a major role [56]. The benzannulation reaction avoids both harsh conditions incompatible with the retention of chiral information and the cumbersome separation of enantiomers, and is thus attractive for the diastereoand enantioselective synthesis of arene complexes [17b, 57].
2708 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
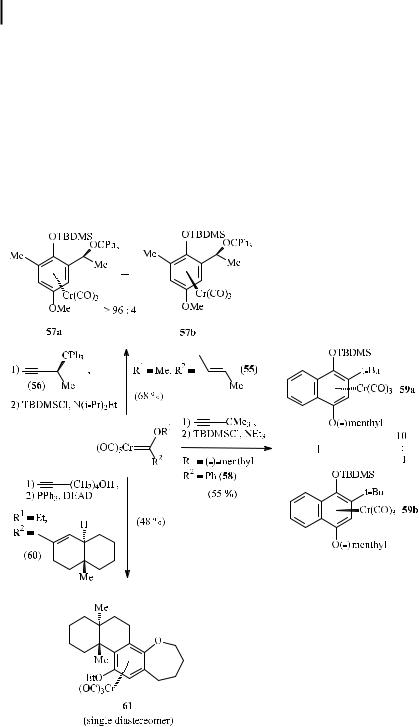
Three di erent strategies have been envisaged. The chiral information can either be incorporated into the alkyne or linked to the heteroatom or to the a,b-unsaturated substituent at the carbene complex carbene carbon. High diastereoselectivities (57a:57b > 96 : 4) have been observed in reactions of vinyl carbene complex 55 with the chiral propargylic ether 56 bearing the bulky trityloxy substituent [57a]. A more general approach is based on chiral alcohols incorporated into the alkoxycarbene complex. Upon benzannulation with tert-butylethyne, the menthyloxy carbene complex 58 gave a diastereoselectivity of 10:1 in favor of the naphthalene tricarbonylchromium complex 59a [57c, 57d]. Finally, the tandem benzannulation– Mitsunobu reaction of optically active carbene complex 60 with 5-hexyn-1-ol a orded the anti-benzoxepine complex 61 as the only diastereomer (Scheme 23) [57b].
Scheme 23. Diastereoselective benzannulation of vinyl and phenyl carbene complexes.
8.3 Scope and Limitations 271
8.3.5
Thermal and Photochemical Benzannulation
The benzannulation reaction is usually performed in ethereal solvents under mild conditions of gentle warming. Modifications of this general protocol involve the use of ultrasound or a dry-state adsorption (DSA) methodology, which involves the adsorption of the reactants onto silica gel, which may speed up the reaction and simplify the work-up procedure in some cases [58a, 58b]. Similarly, photoirradiation of the reaction mixture with light from a xenon lamp has been employed to produce the uncoordinated phenol derivatives without subsequent oxidative work-up [58c].
Stimulated by the photochemical generation of chromium-coordinated ketenes [59d], which have been used in in situ nucleophilic additions and cycloadditions, e orts have been directed towards photoinduced benzannulation. Based on earlier observations of photodecarbonylation processes [59a–c], a photobenzannulation was performed starting from a pre-assembled a,b,g,d-dienyl alkoxy carbene complex [60]. Reacting the alkoxy carbene complex with carbon monoxide, o-methoxyphenols [60a, 60d] were obtained, thus complementing the thermal benzannulation route to p-methoxyphenols. This approach has been extended to the generation of o-aminophenols by reacting dienyl alkoxy carbene complexes with isonitriles [60b, 60c], and dienyl amino carbene complexes with carbon monoxide [60b, 60e]. Examples of this strategy are given in Sections 8.5 and 8.6. Thermal intramolecular benzannulation of dienyl carbene complexes of chromium and tungsten leads to triand tetracyclic benzene derivatives [60f ]. These carbene complexes react with alkynes to give eight-membered carbocycles via 8p-electrocyclization. Benzannulation, however, occurs if the terminal double bond is part of a benzene ring [60g].
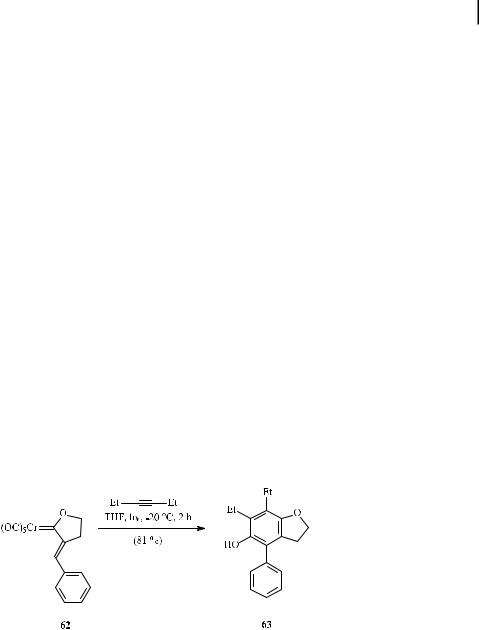
The photochemical protocol may be the method of choice in cases where the thermal reaction fails. This is true for exo-alkylidene oxacyclopentylidene chromium complexes such as 62, which are inert under thermal conditions but undergo a photoinduced benzannulation with, for example, 3-hexyne to give benzofuran 63 (Scheme 24) [61].
Scheme 24. Photochemical benzannulation of an exo-alkylidene-oxacyclopentylidene chromium complex.
8.3.6
Subsequent Transformations
The benzannulation products may be modified by subsequent transformations, by demetalation or by oxidation. To stabilize the coordination of the Cr(CO)3 fragment and thus to re-
2728 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
tain both the stereochemical information based on the plane of chirality and the activation of the arene towards the addition of nucleophiles, it is recommended that the phenolic hydroxy group is protected by silylation or esterification to prevent degradation by oxidation [57c, 57d, 62].
The scope and value of the benzannulation reaction is further increased by the substitution pattern of the arene ring, which can be modified by the incorporation of alkynes bearing additional functional groups such as silyl, stannyl, or boryl substituents. These functional groups have been used in various palladium-catalyzed (cross)-coupling reactions [63, 64]. Further structural elaboration may be based on benzannulation followed by nucleophilic aromatic addition [63b].
A special type of reaction, in which the organometallic moiety changes its coordination to the aromatic p-system, is haptotropic metal migration in polycyclic arene chromium tricarbonyl complexes. The reaction feature is best known for naphthalene chromium tricarbonyl complexes [65]. Since the benzannulation allows a regioselective complexation of polycyclic arenes, the benzannulation products are ideal starting materials for studies of the haptotropic migration. Upon warming the primary (kinetic) benzannulation products, in which the metal fragment is coordinated to the hydroquinoid ring, the metal migrates to the less substituted arene ring to give the thermodynamic regioisomer.
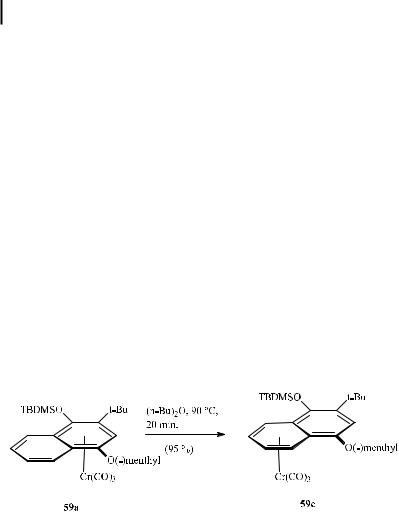
A special application of the haptotropic rearrangement is the reaction of diastereopure complex 59a (Section 8.3.4, Scheme 23). Upon warming to 90 C in di-n-butyl ether, haptotropic migration of the chromium tripod occurs intramolecularly along the same face of the naphthalene system to produce pure diastereomer 59c (Scheme 25) [57d].
Scheme 25. Stereoselective haptotropic rearrangement in naphthalene tricarbonylchromium complexes.
Demetalation of the benzannulation product can be achieved by careful oxidation in air or, more elegantly, by ligand exchange with acetonitrile or carbon monoxide under pressure to give the uncoordinated hydroquinone.
More vigorous oxidation leads directly to quinones. This approach has been employed in a regioselective synthesis of polysubstituted quinones based on benzannulation with trimethylsilyl alkynes followed by oxidation, iododesilylation, and cross-coupling [66].
8.4
Typical Experimental Procedure
The thermal benzannulation is a well-elaborated synthetic technique that provides direct access to Cr(CO)3-activated, densely substituted arenes. While it is possible to obtain the free