

Astruc D. - Modern arene chemistry (2002)(en)
.pdf8.2 Mechanism and Chemoselectivity of the Benzannulation 253
to control the diastereoselectivity of the reaction have met with considerable success. Altogether, after more than two decades, the benzannulation of a,b-unsaturated chromium carbene complexes with alkynes has been developed to a versatile and widely used strategy for the synthesis of densely functionalized monoand polycyclic benzenoid arenes.
The present overview deals with the application of Fischer chromium carbene complexes in the benzannulation reaction for the preparation of highly substituted aromatic compounds. Before focussing on specific arenes (Section 8.5), details of the mechanism are given (Section 8.2), and the scope and limitations of the reaction are defined (Section 8.3). A short description of the experimental procedure is given thereafter (Section 8.4). Finally, the contribution deals with the application of the chromium carbene benzannulation to natural compounds and molecules with biological activity (Section 8.6).
8.2
Mechanism and Chemoselectivity of the Benzannulation
8.2.1
Mechanism
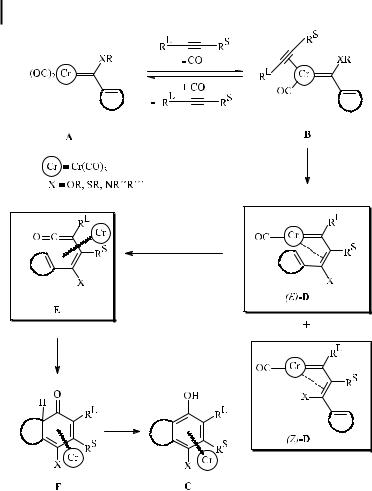
The mechanism of the benzannulation reaction involves the stepwise construction of the aromatic ring within the coordination sphere of the chromium template (Scheme 3). This idea is strongly supported by experimental evidence for model intermediates and by theoretical calculations [9] highlighting the course of the reaction (see below).
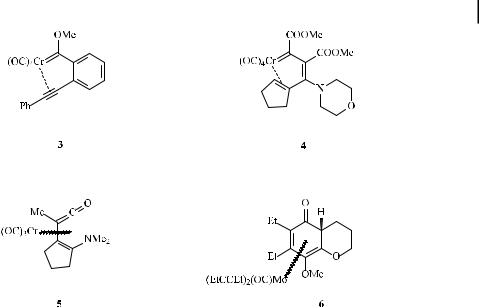
Starting from carbene complex A, ligand exchange of one cis-carbonyl ligand by the alkyne leads to the h2-alkyne complex intermediate B. The equilibrium between A and B is supported by kinetic and inhibition studies under CO atmosphere [10] and has been shown to be the rate-determining step of the benzannulation process. In the course of the decarbonylative ligand exchange, the coordinatively unsaturated 16-electron tetracarbonyl carbene intermediate may be stabilized by the solvent. Recently, based on DFT calculations, an alkyne coordination prior to decarbonylation has been proposed [11a], although this idea is in severe conflict with overwhelming experimental evidence supporting CO dissociation as the first step [11b]. Analogues of intermediate B (linking the alkyne and carbene moieties by a rigid C2-ortho-phenylene) have been isolated as stable alkyne–carbene chromium chelates and structurally characterized by X-ray di raction analysis, which indicated a weak alkyne– metal interaction in complexes of type 3 (Scheme 4) [12]. This finding is in agreement with Extended Hu¨ckel MO studies [13].
Insertion of the alkyne into the chromium carbene bond in intermediate B a ords vinyl carbene complex D, in which the CbC double bond may be either (Z ) or (E ). A putative chromacyclobutene intermediate resulting from a [2þ2] cycloaddition of the alkyne across the metal–carbene bond on the way to chromium vinylcarbene D, as was sometimes suggested in early mechanistic discussions, has been characterized as a high energy species on the basis of theoretical calculations [9c]. Its formation and ring-opening cannot compete with the direct insertion path of the alkyne into the chromium–carbene bond. An example of an (E )-D alkyne insertion product has been isolated as the decarbonylation product of a tetracarbonyl chromahexatriene (4, Scheme 4) [14], and has been characterized by NMR spectroscopy and X-ray analysis.
254 8 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
Scheme 3. Mechanism of the benzannulation reaction (intermediates (E)- and (Z)-D and E serve as branch points, from which side products originate, see Section 8.2.2 for further discussion).
Intramolecular insertion of carbon monoxide into the metal–carbene bond of the (E )- isomer of D leads to the h4-vinyl ketene complex intermediate E. Experimental support for this type of intermediate has been provided by the isolation of Cr(CO)3-coordinated dienyl ketenes related to 5 (Scheme 4) [15a], and by trapping the vinyl ketene intermediates as vinyl lactone derivatives in the course of the reaction of chromium carbene complexes with 1- alkynols [15b].
Electrocyclization of E is expected to give cyclohexadienone complex F; a related molybdenum complex 6 (Scheme 4), in which two carbonyl ligands have been replaced by alkyne ligands, has been isolated from the reaction of a vinyl molybdenum carbene complex with 3-
8.2 Mechanism and Chemoselectivity of the Benzannulation 255
Scheme 4. Isolated analogues of intermediates in the benzannulation reaction.
hexyne [16]. If substituents such as R ¼ H, SiR3 are present in one of the positions adjacent to the carbonyl group in F allowing a 1,3-shift, the cyclohexadienone undergoes tautomerization to phenol C as the final product of the benzannulation sequence. DFT calculations reveal that the CaC bond-forming steps are characterized by rather low activation barriers (10–25 kJ mol 1) and that the thermodynamic driving force for the benzannulation results from the aromatization energy in the final step (ca. 175 kJ mol 1). The tautomerization can be blocked in the presence of ortho-alkyl substituents, and the reaction stops at the cyclohexadienone [17].
The stepwise coupling of two cis ligands as depicted in Scheme 3 has been verified as involving a sequence of three discrete steps at low temperatures, allowing the isolation of the relevant intermediates as individual compounds [18]. When a chelated tetracarbonyl aminovinyl carbene complex (chelated analogue of intermediate B in Scheme 3) was reacted with an electron-deficient alkyne under controlled conditions, a 1,4,5-h3-dienylcarbene tetracarbonyl chromium complex (corresponding to intermediate D in Scheme 3) was formed. It underwent thermal decomposition to give phenol derivatives as the final products.
8.2.2
Chemoselectivity
The intermediate D in Scheme 3 represents a branching point in the benzannulation mechanism, from which side products may arise. Their formation depends on the solvent and on the substitution pattern within the carbene ligand and the alkyne.
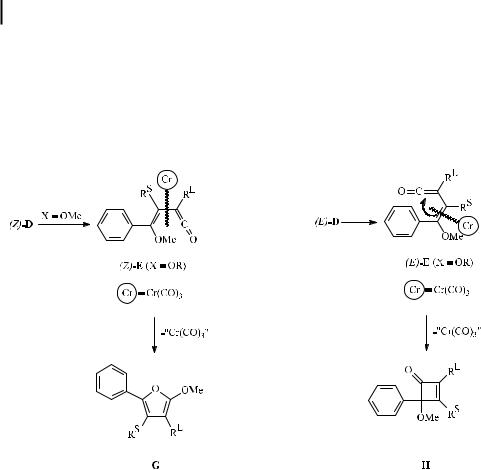
The intermediate D resulting from alkyne insertion may be formed in either the (Z )- or
2568 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
(E )-configuration, which will be retained in the course of subsequent carbonylation to give vinyl ketene complexes (Z )-E or (E )-E. Vinyl ketenes (E )-E provide the appropriate geometry for the 6p-electrocyclization leading to benzannulation. An alternative 4p-electrocyclization, which is favored by internal chelation, strongly coordinating solvents, and o,o0-aryl disubstitution, a ords cyclobutenones H. (Z )-Vinyl ketenes (Z )-E are presumed to be the precursors of furans G, which are believed to result from a cyclization/alkyl migration sequence (Scheme 5) [19].
Scheme 5. Potential side reactions of the benzannulation leading to furans G and cyclobutenones H.
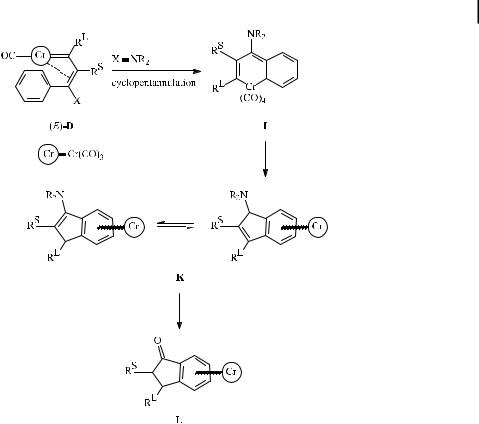
Replacing the alkoxy carbene substituent by a better electron-donating amino group stabilizes the metal carbonyl bond. As a result, CO insertion in vinyl carbene D is hampered; instead, cyclopentannulation via the chromacyclohexadiene I leads to aminoindenes K, which are readily hydrolyzed to indanones L (Scheme 6) [20].
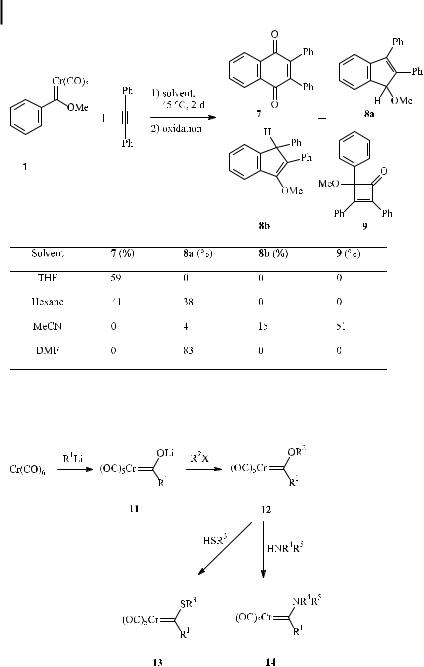
The formation of side products depends on the choice of substituents and solvent [21]. The role of the solvent is illustrated by the reaction of phenyl carbene complex 1 with diphenylethyne (Scheme 7). An ethereal solvent such as THF leads exclusively to the benzannulation product isolated as quinone 7 after oxidative work-up, while use of the noncoordinating solvent hexane results in comparable amounts of cyclobenzannulation and cyclopentannulation products 7 and 8a. Strongly coordinating acetonitrile suppresses benzannulation product 7 in favor of the cyclobutenone 9, which is accompanied by minor amounts of cyclopentannulation products 8a and 8b. Indene 8a is obtained exclusively if the polar solvent DMF is employed.
8.3 Scope and Limitations 257
Scheme 6. Cyclopentannulation of amino carbene complexes.
8.3
Scope and Limitations
8.3.1
The Carbene Complex
8.3.1.1 Availability
Although not yet commercially available, Fischer carbene complexes are readily accessible by well-elaborated synthetic methods.
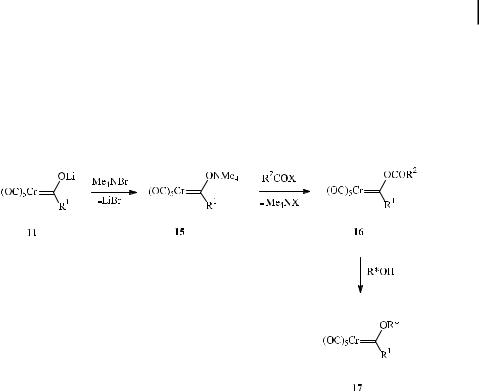
The most general access to Fischer-type metal carbenes (‘‘Fischer route’’) is based on sequential addition of a carbon nucleophile and a carbon electrophile across a metalcoordinated carbon monoxide ligand (Scheme 8) [22]. Addition of an organolithium reagent to hexacarbonyl chromium a ords acyl metalate 11, which undergoes in situ O-alkylation by hard alkylating reagents such as trialkyloxonium tetrafluoroborates or alkyl fluorosulfonates to give alkoxycarbene complex 12 in typical yields of 60–90 % [23, 24]. Alkylation of the acyl metalate has also been performed with methyl iodide under phase-transfer catalysis [25].
258 8 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
Scheme 7. Influence of the solvent on the competition of cyclobenzannulation, cyclopentannulation, and cyclobutenone formation.
Scheme 8. The Fischer route to chromium carbene complexes.
8.3 Scope and Limitations 259
The alkoxy substituent in 12 may be replaced by thio or amino groups upon treatment with thiols and amines to give thiocarbene 13 and aminocarbene complexes 14, respectively.
Alternatively, alkoxycarbene complexes are formed upon alcoholysis of strongly electrophilic acyloxycarbene complexes 16 [26] generated by in situ acylation of tetraalkylammonium acyl metalates 15. These compounds are obtained from lithium precursors 11 and can be stored in a refrigerator for months. This is the method of choice for the synthesis of chiral metal carbenes 17 bearing terpene or sugar auxiliaries (Scheme 9).
Scheme 9. Chiral alkoxy carbene complexes from the alcoholysis of acyloxy carbene complexes.
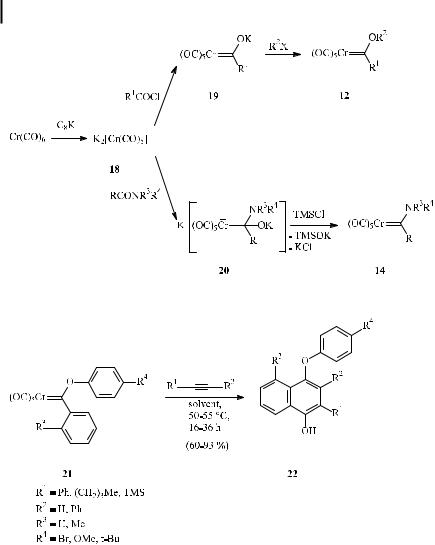
A complementary access to alkoxyand aminocarbene complexes (‘‘Semmelhack– Hegedus route’’) involves the addition of the pentacarbonylchromate dianion 18 (obtained from the reduction of hexacarbonylchromium with C8K) to carboxylic acid chlorides and amides [27] (Scheme 10). While alkylation of acyl chromate 19 leads to alkoxycarbene complexes 12, addition of chromate dianion 18 to carboxylic amides generates the tetrahedral intermediates 20, which are deoxygenated by trimethylsilyl chloride to give amino carbene complexes 14.
8.3.1.2 The Carbene Ligand
Although mostly alkoxy carbene complexes have been benzannulated, other types of carbene complexes are equally well suited. These include aryloxy carbene complexes as well as acylamino and thioalkylidene complexes, and even complexes with no heteroatoms, such as diaryl carbene complexes, are suitable (see below). Besides the commonly used methoxy and ethoxy carbene complexes, alkoxy carbene complexes with a longer alkyl chain have also been successfully reacted. The benzannulation of aryloxy carbene complexes has recently been studied to probe electronic e ects [28a]. Aryloxy alkylidene complexes of type 21 have been used to prepare diaryl ethers 22, which constitute a common substructure in many important types of natural products [28b]. The benzannulation methodology provides an access to phenyl naphthyl ethers in yields of 60–93 % under mild conditions (Scheme 11).
260 8 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
Scheme 10. Semmelhack–Hegedus route to alkoxy and amino carbene complexes.
Scheme 11. Benzannulation of aryloxy carbene complexes a ording diaryl ethers.
Besides alkyloxy carbene complexes, acyloxy carbene complexes have also been proven to be e ective in the benzannulation reaction. For example, Yamashita and Ohishi have reported the synthesis of various naphthoquinones in yields of 47–71 % using acetyloxy phenyl chromium carbene complex and terminal alkynes with di erent alkyl chain lengths [29].
Even Lewis acid adducts of the acyl metalate 11 (Section 8.3.1.1, Scheme 8) proved e ective in generating benzannulation products. The reaction of a trimethylsilyloxy chromium carbene complex with 3-hexyne to give the respective semi-silylated hydroquinone tricarbonylchromium complex in 35 % yield [30a] and the use of titanoxycarbene complexes [30b] constitute such examples. Thiocarbene complexes form hydrothioquinones in a Lewis acid supported benzannulation reaction [32].

8.3 Scope and Limitations 261
As discussed above, dialkylamino carbene complexes result in the formation of indenes due to the increased donor ability of the nitrogen compared to the oxygen heteroatom. The formation of benzannulation products is favored, however, if the electron density at nitrogen is lowered by substitution with electron-withdrawing acyl groups [33]. The example in Scheme 12 demonstrates the e ect.
Scheme 12. Acylamino carbene complexes as amino carbene complex synthons for the benzannulation process.
The facile decarbonylation of pentacarbonyl complexes 23a and 23b results in tetracarbonyl carbene complex intermediates 24a and 24b, respectively. Their annulation can produce benzene and/or cyclopentadiene derivatives 25 and/or 26. In the case of aryl acylamino complex 23a, the reaction course is shifted towards the benzannulation reaction (25a : 26a ¼ 84 : 16). With the vinyl carbene complex 23b, the benzannulation product 25b (25b : 26b ¼ 100 : 0) is produced exclusively.
Recently, Barluenga et al. have introduced some electron-deficient dialkylamino carbene complexes that bear a carboxylic functionality at the vinyl moiety and undergo exclusively the benzannulation process [34].
The benzannulation reaction tolerates phenyl carbene complexes bearing both electrondonating [35a] and -withdrawing [35b] substituents. Additionally, carbene complexes containing condensed systems such as naphthalene and other carbocyclic hydrocarbons as well as heterocycles have also been successfully submitted to the reaction (see Section 8.5).
Representatives of Fischer carbene complexes possessing no heteroatom at the carbene carbon are diaryl chromium carbene complexes. Due to the lack of p-stabilization, they react more readily than the heteroatom-functionalized carbene complexes (already at ambient
2628 The Chromium-Templated Carbene Benzannulation Approach to Densely Functionalized Arenes
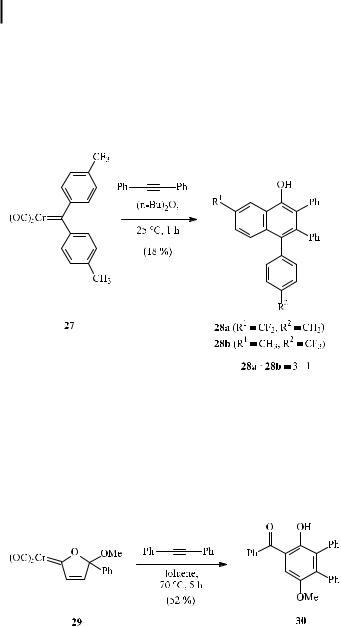
temperature). Diaryl carbene complexes are interesting candidates for synthesizing arenes bearing an extended carbocyclic skeleton [28a, 36]. Scheme 12 shows one example of such a benzannulation [28a, 36a] involving the annulation of a carbene complex bearing two aryl substituents of di erent electron densities. Reaction of compound 27 with diphenylethyne provides a mixture of two regioisomeric benzannulation products 28a and 28b in a 3:1 ratio in a modest yield of 18 % (Scheme 13). Remarkably, the benzannulation occurs preferentially at the aromatic ring having the lower electron density.
Scheme 13. Benzannulation of diaryl chromium carbene complexes.
In contrast to aryl carbene complexes, vinyl carbene complexes are known to yield only the benzannulation products [37]. For instance, carbohydrates [38], tetramethyl ketals of quinones [39], heterocycles, and oxacycloalkenylidene carbene complexes [40] have been used as part of a (cyclic) vinyl carbene complex. For example, complex 29 and diphenylethyne were converted to the acyl hydroquinone 30. Thus, 29 serves as a synthon for the (electron-poor) benzoyl vinyl carbene complex (Scheme 14) [40].
Scheme 14. Complex 29 acting as an acyl vinyl carbene complex synthon in the benzannulation reaction.
The only example of a vinyl carbene complex bearing fluoro substituents that has been benzannulated is presented in Scheme 15 [41]. Carbene complex 31 was reacted with diphenylethyne to give hydroquinone 32. Interestingly, one of the fluoro substituents in the educt is lost in the reaction.