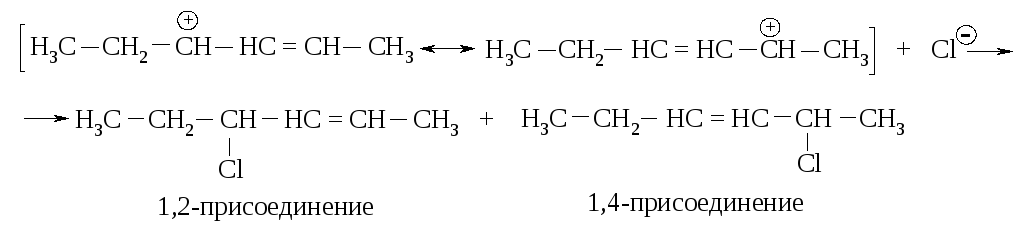9.1. Устойчивость сопряженных диенов
Теплоты гидрирования алкенов одинакового строения близки по значениям. Для монозамещенных алкенов с одной алкильной группой при двойной связи эта величина близка к 126 кДж/моль (табл. 9.1).
Можно было бы ожидать, что для соединений с большим числом двойных связей теплота гидрирования будет равна сумме теплот гидрирования индивидуальных двойных связей. Для несопряженных диенов эта аддитивность соблюдается, теплота гидрирования 1,4-пентадиена 254 кДж/моль (127∙2 кДж/моль). Однако теплота гидрирования 1,3-бутадиена и других сопряженных диенов несколько меньше, чем удвоенная теплота гидрирования связи >С=СН2.
Таблица 9.1
Теплоты гидрирования алкенов и диенов
|
Соединение |
Теплоты гидрирования, кДж/моль |
|
CH3–CH=CH2 (Пропен) |
126 |
|
CH3–CH2–CH2–CH=CH2 (1-Пентен) |
127 |
|
CH2=CH–CH2–CH=CH2 (1,4-Пентадиен) |
254 |
|
CH2=CH–CH=CH2 (1,3-Бутадиен) |
239 |
При гидрировании сопряженного диена экспериментальная теплота гидрирования на 15 кДж/моль меньше, чем рассчитанная по аддитивной схеме. Это означает, что сопряженный диен устойчивее, чем можно было ожидать, исходя из предположения о независимости двойных связей. Выигрыш энергии называется энергией сопряжения.
В чем причина такой устойчивости? Сравним длины простых углерод-углеродных связей, образованных атомами углерода в различных гибридных состояниях (табл. 9.2).
Таблица 9.2
Гибридизация атомов углерода и длины простых углерод-углеродных связей
|
Соединение |
Связь С–С |
Гибридизация |
Длина связи, нм |
|
CH3 C2H2–C3H2–CH3 |
C2–C3 |
sp3– sp3 |
15,410-2 |
|
CH2=C2H–C3H2–CH3 |
C2–C3 |
sp2– sp3 |
15,210-2 |
|
CH2=C2H–C3H=CH2 |
C2–C3 |
sp2– sp2 |
14,810-2 |
Поскольку sp2-орбитали расположены ближе к ядру, чем sp3-орбитали, длина углерод-углеродной связи должна изменяться в следующем порядке: Сsp2–Сsp2 < Сsp3–Сsp2 < Сsp3–Сsp3, что и наблюдается в действительности. Чем короче связь, тем больше ее энергия, т.е. уменьшение длины связи увеличивает устойчивость молекулы.
Вероятно, стабильность сопряженного диена в некоторой степени связана со взаимодействием -электронных облаков соседних -связей, дополнительное электронное взаимодействие уменьшает энергию диена. Возникающая делокализация -электронов делает молекулу более стабильной. На рис. 9.1 показано перекрывание р-орбиталей атомов С2 и С3.
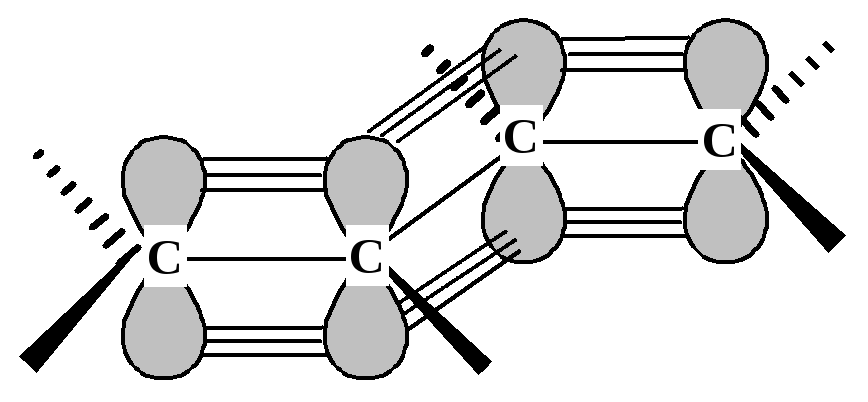
Рис. 9.1. Взаимодействие -электронных облаков в сопряженном диене
Связь С2-С3 не является ни простой, ни двойной. Каков порядок этой связи? Если порядок углерод-углеродной связи для этана - 1, этилена - 2, ацетилена - 3, то порядок для любой связи между атомами углерода может быть определен путем измерения длины этой связи и сравнения ее с длиной связи углерод-углерод в этане, этилене и ацетилене. Этим методом было установлено, что порядок связи C2-C3 в I,3-бутадиене составляет 1,2.
9.2. Химические свойства
9.2.1. Электрофильное присоединение
Галогены, галогенводороды и некоторые другие электрофильные реагенты образуют с сопряженными диенами смесь двух продуктов (1,2- и 1,4-присоединение). В результате реакции 1,3-бутадиена с бромом (20 оС, CCl4) и с бромоводородом (в отсутствие пероксидов, т.е. механизм AdE) реагент присоединяется не только к соседним атомам углерода при двойной связи (1,2-присоединение), но и к концевым атомам сопряженной системы (1,4-присоединение).
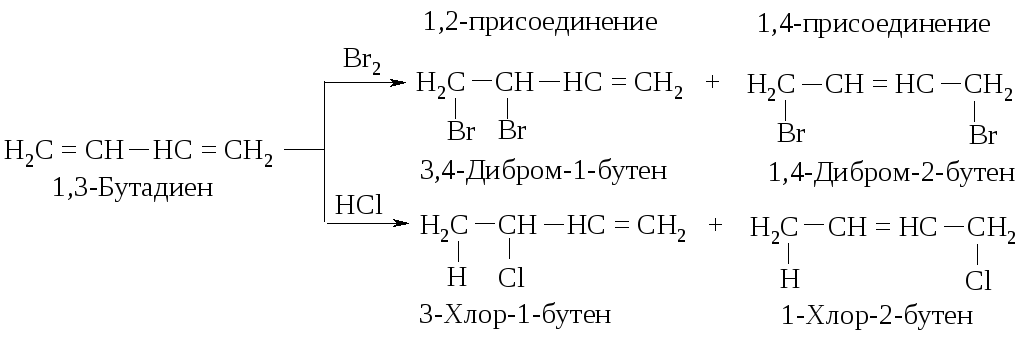
Как объяснить образование таких продуктов? Присоединение хлороводорода и брома к диенам так же, как и присоединение к алкенам, протекает по механизму AdE. Рассмотрим его на примере взаимодействия хлороводорода с 2,4-гексадиеном.
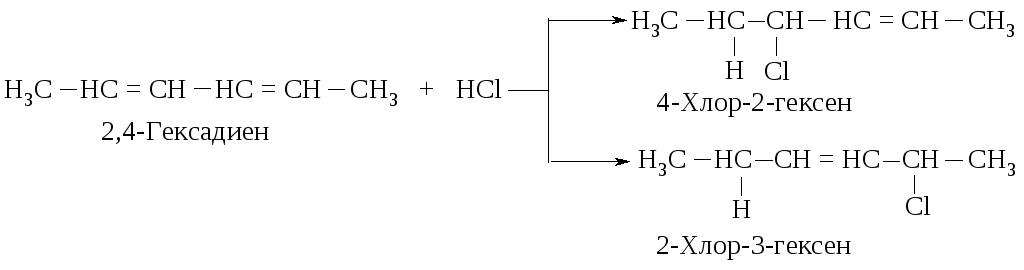
Образование этих продуктов показывает, что первая стадия - присоединение протона - протекает с образованием карбокатиона I, а не II.
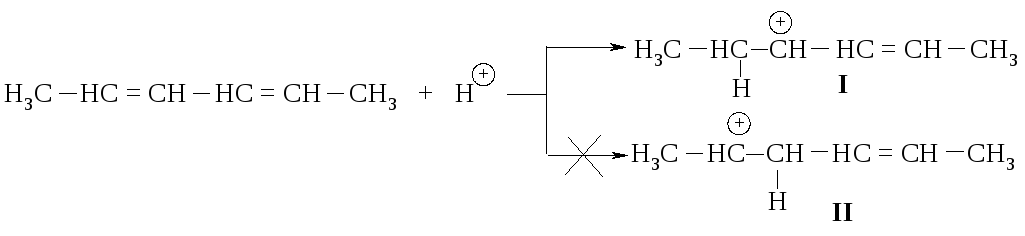
Оба карбокатиона являются вторичными. Почему же предпочтительнее образование иона I? Дело в том, что в ионе I атом углерода, несущий положительный заряд, находится в -положении по отношению к двойной связи, такой карбокатион называется аллильным.
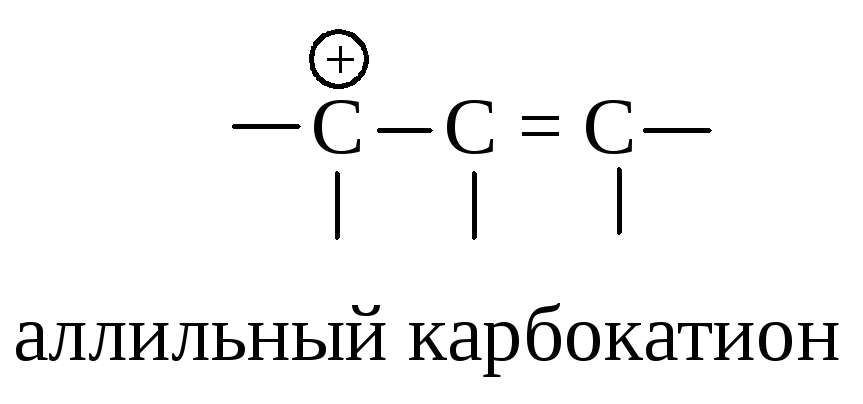
Аллильный карбокатион почти так же устойчив, как третичный карбокатион. Теперь ряд устойчивости карбокатионов можно дополнить аллил-катионом:
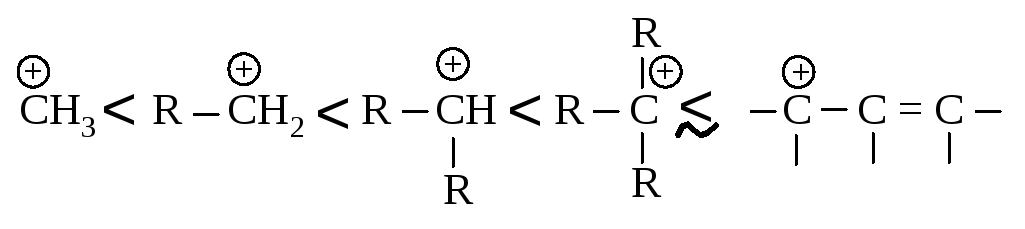
В чем причина устойчивости аллил-катиона? До сих пор мы считали, что молекуле или иону может быть приписана только одна структура. Но аллильный карбокатион I может быть изображен также и структурой III:
![]()
Эти структуры отличаются только положением двойной связи и положительного заряда. Какой же способ изображения правильно показывает распределение электронов? Ни один. В реально существующем аллил-катионе распределение электронов является промежуточным: половина положительного заряда находится на одном атоме углерода и половина - на другом, а связи между этими атомами углерода не являются ни двойными, ни простыми, а промежуточными между ними.
![]()
Изображение реального строения аллил-катиона с помощью только одной структуры с принятыми химиками обозначениями ( - отсутствие электрона, черточка - пара электронов ковалентной связи и т.д.) невозможно. Оно не позволяет представить распределение двух -электронов по р-орбиталям трех атомов углерода.
Для решения подобных проблем химики предложили концепцию резонанса. Основное положение этой теории состоит в следующем: если частица может быть представлена двумя или более структурами, в которых атомные ядра соединены одинаково, а различно только распределение -электронов, то реальное распределение -электронов не может быть удовлетворительно представлено ни одной из них. Реальная молекула представляет собой нечто промежуточное между ними – резонансный гибрид этих структур. Такие гипотетические структуры называют граничными (каноническими). Граничные структуры соединяют знаком ↔.
Граничные структуры должны иметь одинаковое число неспаренных электронов. Структура
![]()
не может быть граничной структурой для аллильного катиона I.
Энергия реальной молекулы (резонансного гибрида) меньше, чем энергия любой из граничных структур. Вклад отдельных канонических структур в истинную структуру частицы неодинаков, наиболее устойчивая структура дает наибольший вклад. Наибольшая стабилизация достигается в тех случаях, когда структуры, вносящие вклад в резонансный гибрид, энергетически эквивалентны. Выигрыш энергии за счет суммирования вкладов всех граничных структур называют энергией резонанса.
Таким образом, в реальном аллил-катионе положительный заряд не локализован на определенном атоме, а распределен между двумя атомами углерода.
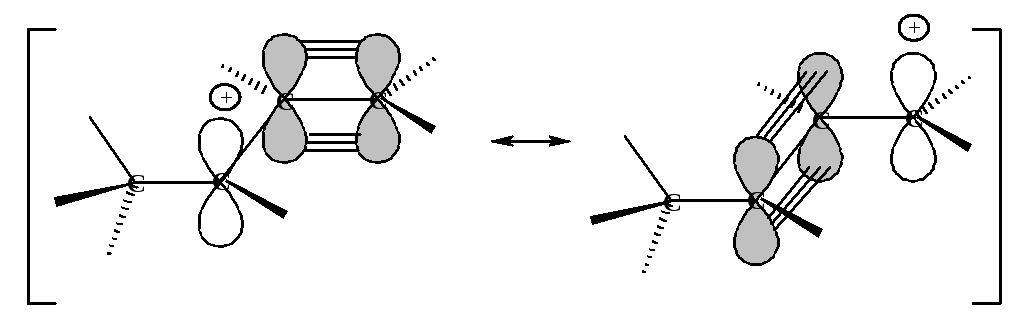
Все атомы, участвующие в резонансе, лежат в одной плоскости, при этом достигается максимальное перекрывание р-орбитали.
Вернемся к механизму реакции хлороводорода с 2,4-гексадиеном. Во второй стадии анион хлора может присоединяться к любому из атомов с положительным зарядом. В результате образуются два продукта.