

5.1.3. Гистидинемия
Гистидинемия (histidinaemia, syndrome Ghadimi-Partington) – наследственное заболевание, в основе которого лежит блок гистидазы (дезаминазы L-гистидина) (cхема 7) – фермента, локализующегося в гепатоцитах и роговом слое кожи. Угнетение активности этого энзима провоцирует накопление в тканях гистидина и продуктов его побочного метаболизма, токсически действующих на ЦНС. Наследуется по аутосомно-рецессивному типу. Частота патологии колеблется от 1:10 000 до 1:100 000.
Гистидиндезаминаза
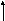
 Гистидин
Уроканиновая
кислота
Гистидин
Уроканиновая
кислота
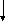
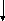
-
Гистидинемия
Имидазолил- Имидазолон-

 пируват
пропионат
пируват
пропионат
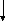
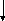
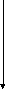 Имидазолил-
Имидазолил- Формиминоглутамат
Имидазолил-
Имидазолил- Формиминоглутамат
ацетат лактат
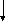
 ТГФК
ТГФК
Имидазолилацетил-
глутамин С1-ТГФК
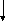
моча Глутамат
Схема 7. Обмен гистидина в норме и при патологии.
Первые симптомы начинают проявляться уже в грудном возрасте, регистрируются мышечная гипотония или судорожный синдром, отставание в психомоторном развитии, позднее возможны из-за кратковременности слуховой памяти замедление разговорных фраз, искажённое произношение слов, предложений. Степень умственной отсталости варьирует в широких пределах (IQ=50–120). В физическом развитии дети не отстают. Характерный фенотипический признак – наличие у пробандов голубых глаз и светлых волос.
В основе лабораторной диагностики лежит выявление гипергистидинемии, высокие цифры аминокислоты в моче (зелёное окрашивание хлоридом железа), но – пониженное содержание уроканиновой кислоты. Иногда используется определение активности гистидазы в клетках кожи.
До настоящего времени основа лечения – диетотерапия. Ограничивают или полностью исключают продукты, богатые гистидином (мясо, яйца, молочную кашу, горох, рис и другие зерновые). Правда, подобный способ коррекции позволяет уменьшить выраженность неврологических симптомов, но мало влияет на интеллект и речь.
5.1.4. Молекулярные болезни обмена триптофана
Синдром Хартнупа (syndrome Hartnup) (схема 8) наследуется по аутосомно-рецессивному типу, редко встречающееся заболевание, но наиболее распространённая форма среди генных повреждений метаболизма триптофана. Получила своё название по фамилии больного, родившегося от двоюродных брата и сестры. Недуг служит классическим примером аминоацидопатий, в основе которых лежит мутация гена, ответственного за синтез белков – переносчиков, обеспечивающих активный транспорт данной аминокислоты, в первую очередь, всасывание в кишечнике и его реабсорбцию в почечных канальцах. В результате развивается дефицит триптофана, особенно необходимого в качестве субстрата в синтезе НАД+ - активной формы витамина РР. Отсюда синдром Хартнупа клинически напоминает пеллагру (экзогенный гиповитаминоз никотинамида), которая обычно включает, как образно, выражаются «синдром 3-х Д»: диарею, дерматит, деменцию. Для данной наследственной патологии характерны нарушения кожи, особенные чувствительные к действию солнечных лучей (фотодерматоз). Неврологические симптомы проявляются психоэмоциональной лабильностью, обморочными состояниями, вероятны атаксия, нистагм. Течение доброкачественное. Возможно самоизлечение.
Диагноз ставят на основании увеличенной экскреции с мочой триптофана, а также других аминокислот (аланина, серина, глутамина) и обнаружения в плазме крови гипотриптофанемии.
Терапия традиционна – диета, обогащённая белками, введение повышенных количеств витаминов РР и В6. В летнее время – защита открытых участков кожи от прямого воздействия солнечных лучей.
Патогенез редкого синдрома «голубых пелёнок» (индиканурии) (схема 8) сходен с развитием синдрома Хартнупа. Разница заключается, в первую очередь, в сроках начала заболевания. Если первый характерен для грудных младенцев, то симптомы второго возникают уже во взрослом состоянии. Вероятным объяснением может служить наличие нескольких генов, ответственных за синтез белков – переносчиков (участников активного транспорта триптофана). Каждый из этих транскриптонов работает в определённый период жизни человека, а другие в это время репрессированы. Отсюда в зависимости от того, какой ген мутирован, патологическое состояние будет возникать или в младенчестве, или позже.
Если нарушена экспрессия гена, функционирующего у новорождённого, триптофан белков грудного молока не всасывается, накапливается в кишечнике, подвергается действию ферментов находящейся в нём микрофлоры. Продукты гниения, в первую очередь, индол всасываются и через воротную вену попадают в печень. В норме данное соединение обезвреживается в гепатоцитах путём конъюгирования: взаимодействует с ФАФС (фосфоаденозинфосфосульфатом), образуется индоксилсерная кислота, которая в виде соли выводится с мочой.