

Глава 5. Наследственная патология азотистого обмена
5.1. Аминоацидопатии
Аминокислоты – продукты гидролиза пищевых белков в ЖКТ; всасываются ворсинками энтероцитов и по портальной системе попадают в печень. Оттуда их основная масса выходит в общий кровоток и доставляется в клетки, где вступает в различные реакции. В отличие от глюкозы и ВЖК, подавляющая часть которых используется как источники энергии, аминокислоты в основном служат кирпичиками в синтезе протеинов тканей. Однако небольшой процент этих соединений подвергается специфическим преобразованиям, в результате которых получаются вещества, необходимые организму.
Находясь в кровеносном русле и обладая низкой молекулярной массой, аминокислоты легко фильтруются в канальцах почек, но с помощью специальных систем активного транспорта частично реабсорбируются.
Отсюда в наследственной патологии данных структур можно выделить три механизма генетических повреждений:
дефект белков – переносчиков, ответственных за активный транспорт в тонком кишечнике (синдром Хартнупа) или в канальцах почек (цистинурия); выделяют следующие дефекты этого механизма для:
а) диаминомонокарбоновых кислот (цистина, орнитина, аргинина,
лизина);
б) моноаминодикарбоновых кислот (аспартата, глутамата);
в) нейтральных аминокислот (аланина, треонина и др.);
г) иминокислот (пролина, гидроксипролина), -
что может проявляться нарушениями экскреции или всасывания
близких по структуре аминокислот;
блок того или иного фермента специфического пути преобразования аминокислоты (гистидинемия);
дефект какого-либо энзима в общих путях метаболизма аминокислот (аргининемия).
5.1.1. Патология синтеза мочевины
Одной из реакций катаболизма аминокислот служит дезаминирование. Высвобождающийся при этом аммиак, легко растворяясь в воде цитозоля, преобразуется в гидроксид аммония – щёлочь, что грозит резким сдвигом рН (алкалозом) - одного из важнейших показателей гомеостаза. Чтобы этого не происходило, на защиту стабильности рН встают буферные системы – образуются соли аммония. Мало того, с целью нейтрализации используются α-кетокислоты, в первую очередь, α-кетоглутарат, который после взаимодействия с двумя молекулами NH3 превращается в глутамин. Последний и соли аммония из клеток током крови доставляются в основном в гепатоциты, где и происходит окончательное обезвреживание аммиака – процесс известный как цикл Кребса – Гензеляйта (орнитиновый цикл). В настоящее время установлены дефекты всех ферментов синтеза мочевины (схема 5).
В большинстве случаев у таких больных отмечаются умственная отсталость, судороги, упорная рвота. Чем на более ранних стадиях тормозится синтез, тем тяжелее течение недуга и его прогноз. Диагноз ставится при регистрации блока соответствующего энзима (табл. 8).
Лечение разработано не до конца. Основной упор делается на диетотерапию: снижение потребления белковых источников, а также введение α-кетоаналогов незаменимых аминокислот, что уменьшает продукцию свободного аммиака; применяют аргинин, цитруллин, орнитин с целью связывания аминогрупп.
Аспартат


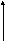
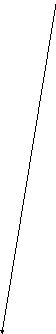 Цитруллин
Аргининосукцинат
Цитруллин
Аргининосукцинат
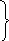
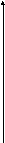 аргининосукцинатсинтаза
аргининосукцинатсинтаза
NH4+,
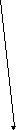
|
Цитруллинемия |
Орнитинкарбомоил-
трансфераза
-
А

 ммониемия
II
ммониемия
II
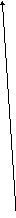 Орнитин
Орнитин
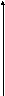

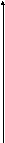 2
АТФ Карбамоилфосфат
Аргинин-
2
АТФ Карбамоилфосфат
Аргинин-
Карбамоилфосфатсинтаза сукцинат-
лиаза
-
А
 ргинин-янтарная
ргинин-янтарнаяацидемия
-
Аммониемия I
Аргиназа
-
А

 ргининемия
ргининемия
Н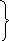 СО3-,
СО3-,
карбокси-
биотин Мочевина
Аргинин
Схема 5. Синтез мочевины и его патология.
Таблица 8