

1)Основные понятия термодинамики
Термодина́мика— раздел физики, изучающий соотношения и превращения теплоты и других форм энергии. В отдельные дисциплины выделились химическая термодинамика, изучающая физико-химические превращения, связанные с выделением или поглощением тепла, а также теплотехника. Термодинамической системой называется тело или группа тел,отделённые от окружающей среды реальной или воображаемой поверхностью раздела. Если между частями системы не существует физических границ границ раздела ,то такие системы называются гомогенными (водный раствор поваренной соли),для гетерогенных сисетм характерно наличие границ (вода с плавающими в ней кусочками льда).С Состояние термодинамической системы определяется совокупностью её физических и химических свойств,характеризующихся экстенсивными и интенсивными параметрами. Состояния системы: -равновесное(свойства постоянны,нет потоков веществ и энергии) -термодинамически обратимое(равновесны все промежуточные стадии) -стандартное. Интенсивные параметры(зависящие от кол-ва вещества в системе- масса объем энергия) Экстенсивные параметры(не зависящие от кол-ва- температура давление плотность). Внутренняя энергия-энергия системы,слагающаяся из энергии кинетического движения,составляющих ее частицы и энергии потенциальной при таком движении.Работа-упорядоченная форма передачи энергии,с изменением параметров состояния системы.Теплота-неупорядоченная форма пердачи. Типы систем: -изолированная(не обменивается ни Е ни m) -закрытая(обмнивается только Е пример ампула) -открытая(обмен и тем и другим пример живая клетка) Процессы: -изотермические(Т постоянная) -изобарные(Р постоянно) -изохорные(vпостоянно).
Вопрос 2Первый закон термодинамики:
приращение внутренней Е сис-мы в некотором процессе = теплоте полученное сис-мой + работаW , совершенная над системой в этом процессе. Дельта E=Q+W.Qобычно отдается в биологич. Системах, поэтому удобно польз. Этой.ф-лой –дельта Е=-Q-W. Е не создается и не уничтож. Возможны лишь ее переходы из одного вида в другой в строго эквивалентных кол-вах. Внутренняя Е: функция состояния, приращение которой= теплотеQполученной сис-мой в изохорном процессе. Тепловой эффект: кол-во теплоты, выделяемой или поглощаемой реагирующей сис-мой. Тепловой эффект=изменению внутренней Е сис-мы при изохорном процессе и изменению энтальпии при изобарном. Энтальпия: функция состояния, приращение которой = теплотеQ, полученное сис-мой в изобарном процессе. дельта Н=дельтаЕ+р*дельтаV. дельтаН сгор характериз ищменения энтальпии происходящее при сгорании 1 моль вещ-ва. Закон Гесса: теплота хим р-ции, протекающей при р илиV=constопределяется только природой исходных в-в и продуктов р-ции и не зависит от числа и последовательности промежуточных стадий при условии, что теплоты измерены при одинаковыхt. 1)теплота образования в-ва=теплоте его разложения, взятой с обратным знаком. 2)энтальпия образования=разности сумм энтальпий образования продукта р-ции и исходных в-в 3)энтальпия сгорания=разности сумм энтальпий сгорания исходных в-в и продуктов р-ции. Термохимическое ур-е: ур-е реакции для которой указываются соответств. этой реакции дельтаЕ и дельтаН или какой либо др. функции состоян В биологических системах термодинамически невыгодные (эндергонические) реакции могут протекать лишь за счёт энергии экзергонических реакций. Экзергонический процесс это самопроизв. протекающ. процесс сопровожд уменьш свободной энергии системы(наприм. Катаболизм) Анаболизм это эндергонический процесс.-Экзергон. процесс передают свободную энергию для осуществл эндеогонич проц-такие как синтез, неспецифич и специфич эффекты возбуждения.В живых кл главным высокоэнергет продуктом явл АТФ.
3.Второе
начало термодинамики ….Второй закон
термодинамики определяет движущую силу
самопроизвольно совершающихся в природе
процессов, в точности хим. реакций путем
рассмотрения еще одной функции состояния
энтропии (S). Самопроизвольный
процесс, проходящий без изменения
энергетического запаса системы,
совершается только в направлении, при
котором система приходит в более
вероятное состояние. Мерой вероятности
данного состояния вещества или системы
является термодинамическая функция
состояния системы – энтропия (S).
Она характеризует меру беспорядка.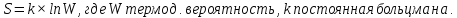
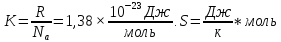 .
.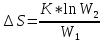 .
Самопроизвольный процесс
.
Самопроизвольный процесс (для изолированных систем). Второй закон
термодинамики: В изолированных системах
самопроизвольно могут совершаться
только такие процессы, при которых
энтропия системы возрастает, энергия
Гиббса уменьшается.. Энтропию можно
рассчитать. Изменение энтропии подчиняется
закону Гесса.
(для изолированных систем). Второй закон
термодинамики: В изолированных системах
самопроизвольно могут совершаться
только такие процессы, при которых
энтропия системы возрастает, энергия
Гиббса уменьшается.. Энтропию можно
рассчитать. Изменение энтропии подчиняется
закону Гесса.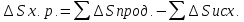 Процесс называется термодинамически
обратимым, если при переходе из начального
состояния в конечное все промежуточные
состояния оказываются равновесными.
Процесс называется термодинамически
необратимым, если хоть одно из промежуточных
состояний неравновесно.Стандартная
эн Гиббса ΔG = ΔH – T ΔS
(G, кДж).
Изолированная сис-ма: не
обменивается со средой ни в-вом, ни Е.Закрытая сис-ма: не обменивается
в-вом, но может обмениваться Е. Пусть ΔН
< 0 (экзотермический процесс), а ТΔS >
0.Тогда из формулы ΔG =ΔН – ТΔS видно, что
ΔG < 0 и процесс протекает самопроизвольно.2).
ΔН> 0 (эндотермический процесс), ТΔS <
0 и тогда ΔG > 0. В этом случае самопроизвольный
процесс не протекает, но может протекать
при затрате энергии из окружающей
среды.3) ΔН = ТΔS и тогда ΔG = 0. Наступает
термодинамическое равновесие в системе.4)
ΔН < 0 и ТΔS < 0, ΔН > 0 и ТΔS > 0. В этом
случае значение ΔG зависит от того, какая
из величин больше: ΔН или ТΔS.Если ТΔS >>
ΔН, то ΔG < 0; если ТΔS << ΔН, то ΔG < 0.
Процесс называется термодинамически
обратимым, если при переходе из начального
состояния в конечное все промежуточные
состояния оказываются равновесными.
Процесс называется термодинамически
необратимым, если хоть одно из промежуточных
состояний неравновесно.Стандартная
эн Гиббса ΔG = ΔH – T ΔS
(G, кДж).
Изолированная сис-ма: не
обменивается со средой ни в-вом, ни Е.Закрытая сис-ма: не обменивается
в-вом, но может обмениваться Е. Пусть ΔН
< 0 (экзотермический процесс), а ТΔS >
0.Тогда из формулы ΔG =ΔН – ТΔS видно, что
ΔG < 0 и процесс протекает самопроизвольно.2).
ΔН> 0 (эндотермический процесс), ТΔS <
0 и тогда ΔG > 0. В этом случае самопроизвольный
процесс не протекает, но может протекать
при затрате энергии из окружающей
среды.3) ΔН = ТΔS и тогда ΔG = 0. Наступает
термодинамическое равновесие в системе.4)
ΔН < 0 и ТΔS < 0, ΔН > 0 и ТΔS > 0. В этом
случае значение ΔG зависит от того, какая
из величин больше: ΔН или ТΔS.Если ТΔS >>
ΔН, то ΔG < 0; если ТΔS << ΔН, то ΔG < 0.
4) а)ХИМИЧЕСКОЕ РАВНОВЕСИЕ – состояние химической системы, при котором возможны реакции, идущие с равными скоростями в противоположных направлениях. При химическом равновесии концентрации реагентов, температура и другие параметры системы не изменяются со временем. б)Обратимые реакции — химические реакции, протекающие одновременно в двух противоположных направлениях (прямом и обратном), например: 3H2 + N2 ? 2NH3.* Направление обратимых реакций зависит от концентраций веществ — участников реакции. Так в приведённой реакции, при малой концентрации аммиака в газовой смеси и больших концентрациях азота и водорода происходит образование аммиака; напротив, при большой концентрации аммиака он разлагается, реакция идёт в обратном направлении. По завершении обратимой реакции, т. е. при достижении химического равновесия, система содержит как исходные вещества, так и продукты реакции.* Простая (одностадийная) обратимая реакция состоит из двух происходящих одновременно элементарных реакций, которые отличаются одна от другой лишь направлением химического превращения. * Необратимые реакции — реакции, при которых взятые вещества нацело превращаются в продукты реакции, не реагирующие между собой при данных условиях, например, разложение взрывчатых веществ, горение углеводородов, образование малодиссоциирующих соединений, выпадение осадка, образование газообразных веществ. Ba(ClO2)2 + H2SO4 > 2HClO2 + BaSO4v NaHCO3 + CH3COOH > CH3COONa + H2O + CO2^ Однако надо понимать, что при изменении условий протекания реакции, теоретически возможно сместить равновесие любой реакции. в) Условия равновесия в процессах переноса теплоты определяют измерениями температур соприкасающихся фаз. Механическое равновесие обнаруживают по равенству непосредственно измеренных давлений в соприкасающихся фазах. Процесс перехода массы из одной фазы в другую в изолированной замкнутой системе, состоящей из двух или большего числа фаз, возникает самопроизвольно и протекает до тех пор, пока между фазами при данных условиях (температуре и давлении) установится подвижное фазовое равновесие. Оно характеризуется тем, что в единицу времени из первой фазы во вторую переходит столько же молекул компонента, сколько из второй в первую. Достигнув состояния равновесия, система может находиться в нем без количественных и качественных изменений сколь угодно долго, пока какое-либо внешнее воздействие не выведет ее из этого состояния. г)Константа химического равновесия — характеристика химической реакции, по значению которой можно судить о направлении процесса при исходном соотношении концентраций реагирующих веществ, о максимально возможном выходе продукта реакции при тех или иных условиях. Константа химического равновесия определяется по закону действующих масс. Ее значения находят расчетно или на основании экспериментальных данных. Константа химического равновесия зависит от природы реагентов и от температуры. д)Положение химического равновесия зависит от следующих параметров реакции: температуры, давления и концентрации. Факторы влияющие на химическое равновесие:1) температура При увеличении температуры химическое равновесие смещается в сторону эндотермической (поглощение) реакции, а при понижении в сторону экзотермической (выделение) реакции. CaCO3=CaO+CO2 -Q t↑ →, t↓ ← N2+3H2↔2NH3 +Q t↑ ←, t↓ → 2) давление При увеличении давления химическое равновесие смещается в сторону меньшего объёма веществ, а при понижении в сторону большего объёма. Этот принцип действует только на газы, т.е. если в реакции участвуют твердые вещества, то они в расчет не берутся. CaCO3=CaO+CO2 P↑ ←, P↓ → 1моль=1моль+1моль 3) концентрация исходных веществ и продуктов реакции При увеличении концентрации одного из исходных веществ химическое равновесие смещается в сторону продуктов реакции, а при понижении концентрации продуктов реакции-в сторону исходных веществ. S2+2O2=2SO2 [S],[O]↑ →, [SO2]↑ ← Катализаторы не влияют на смещение химического равновесия!
5.
Предмет и основные понятия химической
кинетики. Химическая кинетика как основа
для изучения скоростей и механизмов
биохимических процессов. Скорость
реакции. Классификация реакций,
применяющиеся в кинетике: Реакции
гомогенные, гетерогенные; реакции
простые и сложные. Молекулярность
реакции. Кинетические уравнения. Порядок
реакции. Период полупревращения.
Кинетикаизучает скорость химической
реакции, ее зависимость от различных
факторов и механизмы реакций. Изучение
cкорости и механизмов химических
процессов –предмет химической
кинетики. К основным понятиям, на
которых строится теория химических
реакций и химическая кинетика, относятся
такие понятия, как механизм или схема
химической реакции, гомогенность и
гетерогенность, гомофазность и
гетерофазность химической реакции и
реакционного процесса в целом, а также
понятия открытой и замкнутой системы
Важнейшей количественной характеристикой
протекания химической реакции во времени
является скорость реакции. Понятие
скорости реакции характеризует количество
вещества, вступающего в реакцию или
образующегося в результате реакции в
единицу времени. Для гомофазного
химического процесса, идущего при
постоянном объеме, скоростью процесса
по некоторому компоненту называется
изменение концентрации этого компонента
в единицу времени. Если же в процессе
реакции происходит изменение объема
реагирующей системы, то концентрация
вещества оказывается связанной в этом
случае не только с числом актов химического
превращения, но и с тем, по какому закону
изменяется объем системы. В общем случае
это изменение может осуществляться
произвольным образом. Понятие кинетического
уравнения, то есть уравнения, описывающего
зависимость скорости химического
процесса от концентрации компонентов
реакционной смеси. Для подавляющего
большинства химических процессов
скорость реакции может быть представлена
в виде произведения концентрации
реагентов в соответствующих степенях,
называемых порядком реакции по
соответствующему веществу. Еще одним
из важнейших понятий химической кинетики
является понятие энергии активации,
которая характеризует температурную
зависимость скорости химической реакции
в соответствии с законом Аррениуса.
Гомогенность и гетерогенность реакции.
Определяется по агрегатному состоянию
реагентов в (одной фазе и в разных фазах)
Скорость реакции - изменение концентрации
вещества в единицу времени. Ʊ=∆с/∆t.Истинная скорость - положительна .
Ʊист=dс/dtХимическое равновесие - когда
скорости прямой и обратной реакций
сравниваются.Классификация
реакций:1)по механизму: Простые-
осуществляется посредством однотипных
элементарных актов. Сложные- разнотипные
элементарные акты
2) по агрегатному состоянию:
Гомогенные-все исходные вещ-ва в одной
фазе, реакция идет по всему объему смеси
Гетерогенные -
исх. вещ-ва в разных фазах, важна площадь
соприкосновения реагирующих вещ-в.Молекулярность реакции - число
молекул, учавствующих в простой реакции.Кинетические уравнения:
-первого порядка,V=kc(k-константа
скорости реакции)
-второго
порядка,V=kc2
Порядок
реакции- сумма показателей степеней
концентраций реагирующих вещ-в.Период
полупревращений- промежуток времени,
в течении которого начальное кол-во
реагента или его концентрация уменьшаются
в 2 раза.T1/2=ln2/k
Порядок реакции
показывает, каким образом скорость
реакции зависит от концентрации
реагентов. Порядок кинетического
уравнения может принимать значения
0,1,2 и 3; он может быть также дробным.Уравнения нулевого
порядка:С0
– с1=
kt
;C0-0.5
C0=kt0.5
;T0.5= ; Где с0
– начальная концентрация реагента;
; Где с0
– начальная концентрация реагента;
с1 – концентрация реагента в момент времени t;
k – константа скорости реакции;
T0.5 – период полураспада.
Уравнения первого порядка:
ln= =
-kt
;
=
-kt
;
T0.5=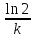 =
=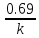 ;
;
Где ct=0.5 c0
Уравнения второго порядка:
 -
-
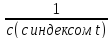 =kt;
=kt;
T0.5=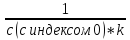 ;
;
6)
Зависимость скорости реакции от
концентрации реагентов. Кинетическое
уравнение реакций первого, второго и
нулевого порядков. Скорость
гомогенной хим. реакции определяется
изменением концентрации реагирующих
веществ (или продуктов реакции) в единицу
времени. Скорость прямой реакции
уменьшается по мере расходования
исходных веществ, а скорость обратной
реакции увеличивается по мере накопления
продуктов реакции. Когда скорости прямой
и обратной реакции сравняются, система
перейдет в состояние химического
равновесия.Истинная скорость в любой
момент времени является только
положительной величиной и определяется
первой производной концентрации по
времени
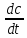 .
Если измерение истинной скорости
проводят по изменению концентрации
исходных веществ, то перед производной
ставят знак минус: V=
-
.
Если измерение истинной скорости
проводят по изменению концентрации
исходных веществ, то перед производной
ставят знак минус: V=
-
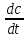 Средняя
скорость:Vср
=
Средняя
скорость:Vср
=
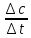
Зависимость скорости хим. реакции от концентрации описывается кинетическим уравнением.
Порядок реакции показывает, каким образом скорость реакции зависит от концентрации реагентов. Порядок кинетического уравнения может принимать значения 0,1,2 и 3; он может быть также дробным.
Уравнения нулевого порядка:
С0 – с1= kt;
C0-0.5 C0=kt0.5 ;
T0.5= ;
;
Где с0 – начальная концентрация реагента;
с1 – концентрация реагента в момент времени t;
k – константа скорости реакции;
T0.5 – период полураспада.
Уравнения первого порядка:
ln= =
-kt
;
=
-kt
;
T0.5= =
=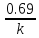 ;
;
Где ct=0.5 c0
Уравнения второго порядка:
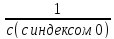 -
-
 =kt;
=kt;
T0.5= ;
;
Закон действующий масс (Закон Гульберта и Вааге): скорость химической реакции пропорциональна произведению концентраций реагирующих веществ в степени их стехиометрических коэффициентов. Т.е. чем больше концентрация, тем больше скорость хим. реакции.
7.
Зависимость скорости реакции от
температуры. Температурный коэффициент
реакции и его особенности для биохимических
процессов. Понятие о теории активных
соударений. Энергетический профиль
реакции; энергия активации; уравнение
Арениуса. Для большинства реакций
справедливо правило Вант-Гоффа: Повышение
температуры на 10К увеличивает скорость
большинства реакций в 2-4 раза:
![]() где V2 — скорость реакции при температуре
Т2, V1— скорость реакции при температуре
Т1, — температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
Из уравнения Вант-Гоффа температурный
коэффициент вычисляется по формуле:
где V2 — скорость реакции при температуре
Т2, V1— скорость реакции при температуре
Т1, — температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
Из уравнения Вант-Гоффа температурный
коэффициент вычисляется по формуле:
![]() Температурный
коэффициент различных реакций различен.
При обычных температурах его значение
для большинства реакций лежит в пределах
от 2 до 4. Это на первый взгляд небольшое
значение температурного коэффициента
обусловливает, однако, большое возрастание
скорости реакции при значительном
повышении температуры. Например, если
температурный коэффициент равен 2,9, то
при возрастании температуры на 100
градусов скорость реакции увеличивается
на 2,9'°, т. е, приблизительно в 50000 раз.
Теория активных соударений обобщает
закономерности зависимости скорости
хим.р-и от температуры: 1.Реагировать
могут не все молекулы, а только находящиеся
в особом активном состоянии 2.Активация
молекулы происходит в результате
биомолекулярного столкновения. 3.При
столкновении частиц с примерно одинаковым
запасом энергии происходит её
перераспределение, в результате чего
энергия одной из молекул достигает
значения, соответствующего энергии
активации. 4.Влияние температуры на
скорость реакции: смещение равновесия
между обычными и активными молекулами
в сторону увеличения концентрации
первых. Энергетический профиль
реакции (график зависимости потенциальной
энергии от координаты реакции)
Температурный
коэффициент различных реакций различен.
При обычных температурах его значение
для большинства реакций лежит в пределах
от 2 до 4. Это на первый взгляд небольшое
значение температурного коэффициента
обусловливает, однако, большое возрастание
скорости реакции при значительном
повышении температуры. Например, если
температурный коэффициент равен 2,9, то
при возрастании температуры на 100
градусов скорость реакции увеличивается
на 2,9'°, т. е, приблизительно в 50000 раз.
Теория активных соударений обобщает
закономерности зависимости скорости
хим.р-и от температуры: 1.Реагировать
могут не все молекулы, а только находящиеся
в особом активном состоянии 2.Активация
молекулы происходит в результате
биомолекулярного столкновения. 3.При
столкновении частиц с примерно одинаковым
запасом энергии происходит её
перераспределение, в результате чего
энергия одной из молекул достигает
значения, соответствующего энергии
активации. 4.Влияние температуры на
скорость реакции: смещение равновесия
между обычными и активными молекулами
в сторону увеличения концентрации
первых. Энергетический профиль
реакции (график зависимости потенциальной
энергии от координаты реакции)
![]() Для
определения скорости и направления
реакции основным фактором является
энергия активации. Скорость любой
химической реакции зависит от числа
столкновений реагирующих молекул в
единицу времени, но если бы все столкновения
сопровождались взаимодействием, то
реакции протекали бы в очень короткие
отрезки времени. Число эффективных
столкновений, по сравнению с числом
реальных столкновений, как правило,
очень мало, что может быть отражено в
уравнении. В 1889 г. С. Аррениус выдвинул
теорию активации, объясняющую сущность
химических реакций. Согласно этой теории
при столкновениях во взаимодействие
вступают только те молекулы, которые
обладают определенным запасом энергии,
необходимой для осуществления той или
иной реакции. Эти молекулы называются
активными молекулами. По-видимому,
скорость химической реакции зависит
от концентрации активных молекул. Это
нашло свое отражение в исследованиях
С. Аррениуса, который показал, что
количество активных молекул может быть
вычислено по уравнению При постоянной
температуре число активных молекул в
среднем сохраняется постоянным.
Минимальный запас энергии, которым
должны обладать молекулы для вступления
в ту или иную реакцию, можно рассматривать
как своеобразный энергетический барьер
этой реакции. Энергия, которая должна
быть сообщена неактивным молекулам для
перехода их в активное состояние,
называется энергией активации. Уравнение
Арениуса устанавливает зависимость
константы скорости химической реакцииkот температуры Т.
Для
определения скорости и направления
реакции основным фактором является
энергия активации. Скорость любой
химической реакции зависит от числа
столкновений реагирующих молекул в
единицу времени, но если бы все столкновения
сопровождались взаимодействием, то
реакции протекали бы в очень короткие
отрезки времени. Число эффективных
столкновений, по сравнению с числом
реальных столкновений, как правило,
очень мало, что может быть отражено в
уравнении. В 1889 г. С. Аррениус выдвинул
теорию активации, объясняющую сущность
химических реакций. Согласно этой теории
при столкновениях во взаимодействие
вступают только те молекулы, которые
обладают определенным запасом энергии,
необходимой для осуществления той или
иной реакции. Эти молекулы называются
активными молекулами. По-видимому,
скорость химической реакции зависит
от концентрации активных молекул. Это
нашло свое отражение в исследованиях
С. Аррениуса, который показал, что
количество активных молекул может быть
вычислено по уравнению При постоянной
температуре число активных молекул в
среднем сохраняется постоянным.
Минимальный запас энергии, которым
должны обладать молекулы для вступления
в ту или иную реакцию, можно рассматривать
как своеобразный энергетический барьер
этой реакции. Энергия, которая должна
быть сообщена неактивным молекулам для
перехода их в активное состояние,
называется энергией активации. Уравнение
Арениуса устанавливает зависимость
константы скорости химической реакцииkот температуры Т.![]() Здесь А характеризует частоту столкновений
реагирующих молекул,R—
универсальная газовая постоянная.
Здесь А характеризует частоту столкновений
реагирующих молекул,R—
универсальная газовая постоянная.
8. Катализ. Гомогенный и гетерогенный катализ. Осбенности каталитич.активности ферментов. Катализ-изменение скорости хим. реакций в присутствии веществ, которые после завершения реакции остаются в неизменном виде и количестве. Увеличение скорости реакции называют положительным катализом, уменьшение – отрицательным катализом (или ингибированием). Катализаторы - вещества, вызывающие положительный катализ; ингибиторы – вещества, замедляющие реакции. Катализаторы ускоряют только термодинамически возможные реакции! Различают гомогенный и гетерогенный катализ. Если катализатор образует одну фазу с реакционной смесью, то такой катализ называют гомогенным (пример- реакцияH2O2в водном растворе в присутствии дихромат-ионов). Если катализатор находится в разных фазах с реакционной смесью, то такой катализ называют гетерогенным (пример - H2O2в водном растворе (жидкая фаза) в присутствииMnO2 (твердая фаза) Катализатор участвует в химической реакции, образуя промежуточные соединения. Катализаторы биохимических реакций имеют белковую природу и называются ферментами. Ферменты несколько отличаются от обычных катализаторов: 1) обладают значительно более высокой каталитической эффективностью; 2)высокая специфичность, т.е. избирательность действия; 3) многие ферменты проявляют каталитическую активность только по отношению к одному субстрату; 4)ферменты проявляют максимальную эффективность только в мягких условиях, характеризующихся небольшим интервалом температур и значений рН. Снижение активности фермента при температуре выше оптимальной связано с тепловой денатурацией белка, которая наступает при 50-60, а иногда и 400C Активность фермента равна скорости реакции нулевого порядка.
9. Роль воды и растворов в жизнедеятельности. Термодинамика растворения. Раствор-это гомогенная система переменного состава из двух и более веществ, находящаяся в состоянии равновесия.Классификация:1) взвеси(грубо-дисперсная система): суспензии(тв.в-во в жидкости) и эмульсии(жидк. в жидк.)2)коллоиды, золи(тонко-дисперсные системы).Значение растворов в жизнедеятельности: многие хим.процессы протекают лишь при условии, что участвующие в них вещества находятся в растворенном состоянии. Важнейшие биологические жидкости(кровь, лимфа, моча, слюна, пот) являются растворами солей, белков, углеводов, липидов в воде. Усвоение пищи связано с переходом питат.веществ в растворенное состояние. Биохимические реакции в живых организмах протекают в растворах. Биожидкости участвуют в транспорте питат.веществ(жиров, аминокислот, кислорода), лекарственных препаратов к органам и тканям, а также в выведении из организма метаболитов. В жидких средах организма поддерживается постоянство кислотности, концентрации солей и органических веществ (концентрационный гомеостаз). Самым распространенным растворителем на нашей планете являетсявода. Особенности воды: по своей теплоемкости превосходит все вещества; аномальное поведение при охлаждении – вода уплотняется, начинает тонуть, потом поднимается(все др.вещества тонут при уплотнении); может возгоняться(возгонка воды) – сублимация(при определен.условиях лед может переходить в пар без предварительного превращения в жидкую воду, т.е. без плавления); вода растворяет все вещества(вопрос только сколько?); высокая диэлектрическая постоянная воды(величина, показывающая во сколько раз сила взаимодействия между двумя зарядами в данном веществе меньше, чем в вакууме); высокая критическая температура; вода является амфолитом(не кислота, не осн-е); участвует в создании полимерных структур организма(белок, липиды…); основа мембранного транспорта.Термодинамика растворения: согласно 2-му началу термодинамики прир, Т=const вещества самопроизвольно могут растворяться в каком-либо растворителе, если в результате этого процесса энергия Гиббса системы уменьшается, т.е. ∆G=(∆H - T∆S)<0. (∆H-энтальпийный фактор, T∆S-энтропийный фактор растворения). При растворении жидких и твердых веществ∆S>0. При растворении газов в жидкости∆S<0. Изменение энтальпии представляет собой алгебраическую сумму изменения энтальпии∆Hкр в результате разрушения кристаллической решетки и изменения энтальпии∆Hсол за счет сольватации частицами растворителя∆Hраств= ∆Hкр + ∆Hсол . При растворении газов энтальпия∆Hкр=0, т.к. не надо затрачивать энергию на разрушение кристаллической решетки. При растворении может происходить изменение и энтропии, и энтальпии.
10.
Понятие об идеальном растворе. Константа
растворимости, условия образования и
растворения осадка. Идеальный
раствор- это
разбавленный раствор неэлектролитов,
который подчиняется законам Вант-Гоффа
и Рауля. Энтальпия смешивания равна 0
(гомогенные смеси углеводородов;
гипотетический раствор, где равенство
всех сил межмолекулярного взаимодействия.)
Произведение
растворимости (ПР,
Ksp) —
произведение концентраций ионов
малорастворимого электролитав
его насыщенном
растворепри
постоянной температуре и давлении.
Произведение растворимости — величина
постоянная. При
постоянной температуре в насыщенных
водных растворах малорастворимых
электролитов устанавливается равновесиемежду
твердым веществом и ионами, образующими
это вещество. Например, в случае для
CaCO3 это
равновесие можно записать в виде:
![]() Константа
этого равновесия рассчитывается по
уравнению:
Константа
этого равновесия рассчитывается по
уравнению:![]() В
приближении идеального
растворас
учётом того, что активностьчистого
компонента равна единице, уравнение
упрощается до выражения:
В
приближении идеального
растворас
учётом того, что активностьчистого
компонента равна единице, уравнение
упрощается до выражения:
![]() Константа
равновесиятакого
процесса называется произведением
растворимости.
В
общем виде, произведение растворимости
для вещества с формулой AmBn,
которое диссоциирует на m ионов An+ и
n ионов Bm-,
рассчитывается по уравнению:
Константа
равновесиятакого
процесса называется произведением
растворимости.
В
общем виде, произведение растворимости
для вещества с формулой AmBn,
которое диссоциирует на m ионов An+ и
n ионов Bm-,
рассчитывается по уравнению:
![]() где
[An+]
и [Bm-] —
равновесные молярные концентрации
ионов, образующихся при электролитической
диссоциации.
Из
произведений растворимости можно
рассчитать концентрации катионов и
анионов в растворе малорастворимого
электролита. Значения произведений
растворимости приведены в справочниках.
Условия растворения
и образования осадков
1)Электролит
выпадет в осадок, если произведение
концентрации его ионов в растворе больше
константы растворимости с(Ba)*с(CO3)>Kпр[BaCO3]
2)Осадок его растворится если все
наоборот
где
[An+]
и [Bm-] —
равновесные молярные концентрации
ионов, образующихся при электролитической
диссоциации.
Из
произведений растворимости можно
рассчитать концентрации катионов и
анионов в растворе малорастворимого
электролита. Значения произведений
растворимости приведены в справочниках.
Условия растворения
и образования осадков
1)Электролит
выпадет в осадок, если произведение
концентрации его ионов в растворе больше
константы растворимости с(Ba)*с(CO3)>Kпр[BaCO3]
2)Осадок его растворится если все
наоборот
11. Коллигативные свойства разбавленных растворов электролитов и неэлектролитов. Закон Рауля и следствия из него: понижение температуры кристаллизации, повышение температуры кипения растворов, осмос. Осмотическое давление, закон Вант-Гоффа. Осмотическое давление растворов электролитов. Гипо-, гипер- и изотонические растворы. Изотонический коэффициент. Понятие об изоосмии (электролитном гомеостазе). Роль осмоса в биологических системах. Плазмолиз и гемолиз. Коллигативные свойства растворов – такие их свойства, которые зависят в основном от концентрации растворенных частиц и гораздо меньше от их размера, молярной массы и других свойств. - понижение давления пара - повышение температуры кипения - понижение температуры замерзания - осмотическое давлениеЗакон Рауля: Пар, находящийся в равновесии с жидкостью, называют насыщенным. Давление такого пара над чистым растворителем (p0) называют давлением или упругостью насыщенного пара чистого растворителя. Давление пара раствора, содержащего нелетучее растворенное вещество, прямо пропорционально мольной доле растворителя в данном растворе (другая формулировка: относительное понижение давления насыщенного пара растворителя над раствором нелетучего электролита равно молярной доле растворенного вещества).p = p0* Х1, где p — давление насыщенного пара растворителя над раствором; p0— давление насыщенного пара над чистым растворителем; Х1 —мольная доля растворителя. p0-p=∆p – понижение давления насыщенного пара растворителя над раствором ∆p/p0=Х2, Х1+Х2=1 Для растворов электролитов используют несколько другую форму уравнения, позволяющую добавить в неё изотонический коэффициент:∆p/p0=i*Х2, где Δp — собственно изменение давления по сравнению с чистым растворителем; Х2— мольная доля вещества в растворе,i– изотонический коэффициент, показывающий во сколько раз экспериментально найденная величина больше теоретической. Следствия: 1. температура кипения раствора выше температуры кипения растворителя. Это обусловлено тем, что давление насыщенного пара растворителя над раствором становится равным атмосферному давлению (условие кипения жидкости) при более высокой температуре, чем в случае чистого растворителя. Повышение температуры кипения Ткип пропорционально моляльности раствора ..∆Ткип=Kэб*Сm, где Кэб – эбулиоскопическая константа растворителя, Сm – моляльность. . Сm=m (раств в-ва)*1000/М (раств в-ва)*q ( масса раст-ля) [моль/кг] 2,температура замерзания (кристаллизации) раствора ниже температуры замерзания (кристаллизации) чистого растворителя. Условием кристаллизации является равенство давления насыщенного пара растворителя над раствором давлению пара над твёрдым растворителем. Поскольку давление пара растворителя над раствором всегда ниже, чем над чистым растворителем, это равенство всегда будет достигаться при температуре более низкой, чем температура замерзания растворителя. Разность между температурой кристаллизации растворителя и температурой начала кристаллизации раствора есть понижение температуры кристаллизации. Понижение температуры замерзания (кристаллизации) Тзам пропорционально моляльности раствора ∆Тзам=Kкр*Сm, где Ккр – криоскопическая константа растворителя. Осмос - самопроизвольный переход растворителя через полупроницаемую мембрану, разделяющую раствор и растворитель или два раствора с различной концентрацией растворенного вещества. Осмос обусловлен диффузией молекул растворителя через полупроницаемую перегородку, которая пропускает только молекулы растворителя. Молекулы растворителя переходят из растворителя в раствор или из менее концентрированного раствора в более концентрированный. Осмотическое давление– сила, обусловливающая осмос (величина, измеряемая минимальным гидравлическим давлением, которое нужно приложить к раствору, чтобы осмос прекратился). Оно равно внешнему давлению, при котором осмос видимо прекращается. За счет осмотического давления сила заставляет жидкость подниматься вверх, это осмотическое давление уравновешивается гидростатическим давлением. Когда скорости диффундирующих веществ станут равны, тогда осмос прекратится. Осмотическое давление возрастает с увеличением концентрации растворенного вещества и температуры и не зависит ни от природы растворенного вещества, ни от природы растворителя. Закон Вант-Гоффа. Вант-Гоффпредположил, что для осмотического давления можно применить уравнение состояния идеального газа:pV = nRТ, илиp=(n/V)RТоткуда Pосм = сRТ, где p - осмотическое давление (кПа), с – молярная концентрация раствора (моль/л) илиPосм=CmRT, гдеCm– моляльность (моль/кг),R=0,082 [л*А*о/К*моль]. Осмотическое давление растворов электролитов. Pосм=i*Cm*R*T,гдеi– изотонический коэффициент (i=α *(n-1)+1, α – степень диссоциации,n– число ионов, обр-ся в результате диссоциации) Гипо-, гипер- и изотонические растворы. Растворы с одинаковым осмотическим давлением называютсяизотоническими. При помещении клеток в изотонический раствор они сохраняют свой размер и нормально функционируют. Плазма крови изотонична физрастворуw(NaCl)=0,86%.Раствор, имеющий более высокое осмотическое давление по сравнению с другим раствором, называетсягипертоническим, имеющий более низкое –гипотоническим. Изотонический коэффициент. i– изотонический коэффициент, показывающий во сколько раз экспериментально найденная величина больше теоретической. i=α *(n-1)+1, α – степень диссоциации,n– число ионов, обр-ся в результате диссоциации. i=∑всех частиц/исходное число раств молекул. i=∆P’/∆P=∆P’осм/∆Pосм=∆T’зам/∆Tзам=∆T’кип/∆Tкип, где значения со штрихом – растворы электролитов, в знаменателях – теорет вычисления. Изоосмия - относительное постоянство осмотического давления в жидких средах и тканях организма, обусловленное поддержанием на данном уровне концентраций содержащихся в них веществ: электролитов, белков. Это одна из важнейших физиологических констант организма, обеспечиваемых механизмами саморегуляции (Гомеостаз) Роль осмоса в биологических системах. Явление осмоса играет важную роль во многих химических и биологических системах. Благодаря осмосу регулируется поступление воды в клетки и межклеточные структуры. Упругость клеток (тургор), обеспечивающая эластичность тканей и сохранение определенной формы органов, обусловлена осмотическим давлением. Животные и растительные клетки имеют оболочки или поверхностный слой протоплазмы, обладающие свойствами полупроницаемых мембран. Осмос участвует в переносе питательных веществ в стволах высоких деревьев, где капиллярный перенос не способен выполнить эту функцию. Плазмолиз - при помещении клеток в гипертонический раствор вода из клеток уходит в более концентрированный раствор и наблюдается сморщивание клеток. Гемолиз- разрушение эритроцитов, сопровождающееся выходом из них гемоглобина. Если клетки красных кровяных телец поместить в гипотонический р-р, то выравнивание давления происходит за счет перехода воды из р-ра в клетку. Клетки набухают и лопаются.
12. Элементы теории растворов электролитов. Сильные и слабые электролиты. Константа ионизации слабого электролита. Закон разведения Оствальда. Ионная сила раствора. Активность и коэффициент активности ионов. Электролиты в организме, слюна как электролит.
Электролиты– это вещества с ионными или сильнополярными ковалентными связями в водных растворах, подвергающиеся электролитической диссоциации, в результат чего образуются катионы и анионы.
Сильные электролиты– вещества, способные диссоциировать нацело. К ним относится большинство солей, а так же некоторые вещества молекулярного строения (HCl).
Слабые электролитыдиссоциируют в незначительно степени, и преобладающей формой их является молекулярная (H2S, органические кислоты). Количественно способность молекулярного электролита к диссоциации определяетсястепенью ионизации(она зависит от концентрации электролита):
α=
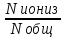 , гдеNобщ – общее число
молекул в растворе;Nиониз
– число молекул, распавшихся на ионы.
, гдеNобщ – общее число
молекул в растворе;Nиониз
– число молекул, распавшихся на ионы.
Константа ионизации:Kd=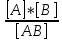 , Где [A],[B]
– распавшиеся ионы [AB] –
не распавшееся на ионы вещество.
, Где [A],[B]
– распавшиеся ионы [AB] –
не распавшееся на ионы вещество.
Закон разбавления Оствальда:
K= α2c/1- α , Где α – степень ионизации С – молярная концентрация
Ионная сила раствора:I=0.5∑сizi2, Где сi – молярная концентрация иона в растворе, моль/л,zi – заряд иона.
Активность иона– это его эффективная концентрация. Активность связана с молярной концентрацией следующим образом:a=f*c, гдеf–коэффициент активности
Электролиты в организме: Na и Clучаствуют в поддержании кислотно-щелочного баланса, осмотического равновесия в организме. Саиграет большую роль в построении костной ткани и зубов, в регулировании кислотности крови и ее свертывании, в возбудимости мышечной и нервной ткани.Кнаходится преимущественно в жидкостях тела и мягких тканях, где является необходимым элементом для поддержания осмотического давления, регуляции рН крови. Mg является кофактором многих ферментативных реакций, необходим на всех этапах синтеза белка. В живых организмах Fe является важным микроэлементом, катализирующим процессы обмена кислородом. Сo входит в состав витамина В12, задействован при кроветворении, функциях нервной системы и печени, ферментативных реакциях. Zn необходим для метаболизма витамина E, участвует в синтезе разных анаболических гормонов в организме, включая инсулин, тестостерон и гормон роста. Mn оказывает влияние на рост, образование крови и функции половых желёз. Слюна как электролит является сложной биохимической средой. Количество ионов Н+ и ОН" определяет рН слюны, который в норме равен 6,9. Величина водородного показателя изменяется в зависимости от характера патологического процесса в полости рта. Так. при инфекционных заболеваниях реакция слюны кислая. Из неорганических веществ в слюне содержатся анионы хлора, брома, иода, фтора. Анионы фосфатов, фтора способствуют увеличению электрохимических потенциалов, анион хлора - переносу ионных зарядов и является деполяризатором (фактор, ускоряющий анодные и катодные процессы). В слюне определяются микроэлементы: железо, медь, серебро, марганец, алюминий и др. - и макроэлементы: кальций, калий, натрий, магний, фосфор.
13.Основные положения протолитической теории кислот и оснований Бренстеда-Лоури;Сопряженная протолитическая пара,амфолиты.Теория Льюиса.Теория Бренстеда-Лоури: кислотойназывают всякое вещество, молекулярные частицы которого(в том числе и ионы) способны отдавать протон, т.е. бытьдонором протонов;основаниемназывают всякое вещество, молекулярные частицы которого(в том числе и ионы) способны присоединить протоны, т.е. бытьакцептором протонов. Например: HNO3 + H2O= H3O++ NO3- Одно и то же вещество в зависимости от условий взаимодействия может быть как кислотой, так и основанием (амфотерность). Например, вода при взаимодействии с сильными кислотами является основанием: H2O + H+ = H3О+, а реагируя с аммиаком, становится кислотой: NH3 + H2O=NH4+ + OH−.Молекула и ион, отличающиеся по составу на один протон, называютсясопряженной кислотно-основной парой. Частицы, способные к взаимодействию как с кислотами, так и с основаниями, называютсяамфолитами. Теория Льюиса:кислотойназывают вещество, принимающие электронные пары, -акцептор электронов;основаниемназывают вещество, поставляющее электроны для образования химической связи, -донор электронов. Например: NH3 + HCI=NH4CI PCl5 + Cl− → PCl6-
14.
Автопротолиз
–обратимый процесс образования равного
числа катионов и анионов из незаряженных
молекул жидкого индивидуального вещества
за счет передачи протона от одной
молекулы к другой. H2O + H2O= H3O+ + OH–.
Это равновесие называется равновесием
автопротолиза воды.
Константа
автопротолиза для воды
обычно называется ионным произведением
воды и обозначается как Kw. Ионное
произведение численно равно произведению
равновесных концентраций ионов
гидроксония и гидроксид-анионов. Обычно
используется упрощенная запись:![]() При стандартных условиях ионное
произведение воды равно 10-14.
Оно является постоянной не только для
чистой воды, но также и для разбавленных
водных растворов веществ. Автопротолиз
воды объясняет, почему чистая вода, хоть
и плохо, но всё же проводит электрический
ток. pH
— это водородный
показатель—
мера активности (в случае разбавленных
растворов отражает концентрацию) ионов
водорода в растворе, количественно
выражающая его кислотность, вычисляется
как отрицательный (взятый с обратным
знаком) десятичный логарифм концентрации
водородных ионов, выраженной в молях
на литр: pH = -log[H+]. Т.е. рН определяется
количественным соотношением в воде
ионов Н+ и ОН-, образующихся при диссоциации
воды. (Моль — единица измерения количества
вещества.) Если
в воде пониженное содержание свободных
ионов водорода
[H+] (рН > 7) по сравнению с ионами гидроксида
[ОН-], то вода будет иметь щелочную
реакцию, а при повышенном содержании
ионов Н+ (рН < 7) - кислую. В идеально
чистой дистиллированной воде эти ионы
будут уравновешивать друг друга и в
нейтральной воде рН=7. При растворении
в воде различных химических веществ
этот баланс может быть нарушен, что
приводит к изменению значения рН. Когда
концентрации обоих видов ионов в растворе
одинаковы, говорят, что раствор имеет
нейтральную реакцию.
При стандартных условиях ионное
произведение воды равно 10-14.
Оно является постоянной не только для
чистой воды, но также и для разбавленных
водных растворов веществ. Автопротолиз
воды объясняет, почему чистая вода, хоть
и плохо, но всё же проводит электрический
ток. pH
— это водородный
показатель—
мера активности (в случае разбавленных
растворов отражает концентрацию) ионов
водорода в растворе, количественно
выражающая его кислотность, вычисляется
как отрицательный (взятый с обратным
знаком) десятичный логарифм концентрации
водородных ионов, выраженной в молях
на литр: pH = -log[H+]. Т.е. рН определяется
количественным соотношением в воде
ионов Н+ и ОН-, образующихся при диссоциации
воды. (Моль — единица измерения количества
вещества.) Если
в воде пониженное содержание свободных
ионов водорода
[H+] (рН > 7) по сравнению с ионами гидроксида
[ОН-], то вода будет иметь щелочную
реакцию, а при повышенном содержании
ионов Н+ (рН < 7) - кислую. В идеально
чистой дистиллированной воде эти ионы
будут уравновешивать друг друга и в
нейтральной воде рН=7. При растворении
в воде различных химических веществ
этот баланс может быть нарушен, что
приводит к изменению значения рН. Когда
концентрации обоих видов ионов в растворе
одинаковы, говорят, что раствор имеет
нейтральную реакцию.
15. Типы протолитических реакций. Гидролтиз. К протолитическим реакциямотносят химические процессы, суть которых заключается в переносе протона от одних реагирующих веществ к другим. протолитическая теория кислот и оснований, в соответствии с которой кислотой считают любое вещество, отдающее протон, а основанием - вещество, способное присоединять протон, например:
CH3COOH + H2O = CH3COO- + H3O+
кислотаI основаниеI основаниеI кислотаII
NH3 + H2O = NH4+ + OH-
основаниеIкислотаII
кислотаII основаниеI К протолитическим
реакциям относят реакции нейтрализации
и гидролиза. Реакция 1 типауксусная кислота с водой:![]() протекающая в прямом
направлении,представляет ионизацию
уксксной кислот,в обратном
направлении-нейтрализацию какого –либо
ацетета. Реакция 2 типаNH4
+H2O=NH3+H3Oпротекает в прямом направлении показывает
гидролиз какой-либо соли аммония ,а в
обратном направлении-нейтрализацию
аммиакаРеакциИ 3типа
протекающая в прямом
направлении,представляет ионизацию
уксксной кислот,в обратном
направлении-нейтрализацию какого –либо
ацетета. Реакция 2 типаNH4
+H2O=NH3+H3Oпротекает в прямом направлении показывает
гидролиз какой-либо соли аммония ,а в
обратном направлении-нейтрализацию
аммиакаРеакциИ 3типа![]() имеют место не только в воде но ив других
растворителях например в жидком аммиаке.Гидро́лиз— один из видов химических
реакций сольволиза, где при взаимодействии
веществ с водой происходит разложение
исходного вещества с образованием новых
соединений. Механизм гидролиза соединений
различных классов: соли, углеводы, белки,
сложные эфиры, жиры и др. имеет существенные
различия. гидролиз жиров, белков и
углеводов происходит при переваривании
пищи, а при гидролизе АТФ выделяется
энергия, обеспечивающая нужды клетки.При
гидролизе солей вода является источником
протонов и электронов. Алкалиметрия
иацидиметрия— важнейшие
титриметрические методы определения
кислот или же оснований, основанные на
реакции нейтрализации:Н+ + ОН− =
Н2ОТитрование раствором щелочи называетсяалкалиметрией, а титрование раствором
кислоты —ацидиметрией
имеют место не только в воде но ив других
растворителях например в жидком аммиаке.Гидро́лиз— один из видов химических
реакций сольволиза, где при взаимодействии
веществ с водой происходит разложение
исходного вещества с образованием новых
соединений. Механизм гидролиза соединений
различных классов: соли, углеводы, белки,
сложные эфиры, жиры и др. имеет существенные
различия. гидролиз жиров, белков и
углеводов происходит при переваривании
пищи, а при гидролизе АТФ выделяется
энергия, обеспечивающая нужды клетки.При
гидролизе солей вода является источником
протонов и электронов. Алкалиметрия
иацидиметрия— важнейшие
титриметрические методы определения
кислот или же оснований, основанные на
реакции нейтрализации:Н+ + ОН− =
Н2ОТитрование раствором щелочи называетсяалкалиметрией, а титрование раствором
кислоты —ацидиметрией
16. Буферными называют растворы, рН которых практически на изменяется от добавления к ним небольших количеств сильной кислоты или щелочи, а также при разведении. Простейший буферный раствор – это смесь слабой кислоты и соли, имеющей с этой кислотой общий анион (например, смесь уксусной кислоты СН3СООН и ацетата натрия СН3СООNa), либо смесь слабого основания и соли, имеющей с этим основанием общий катион (например, смесь гидроксида аммония NH4OH с хлоридом аммония NH4Cl).
С точки зрения протонной теории1 буферное действие растворов обусловлено наличием кислотно-основного равновесия общего типа: Воснование + Н+ Û ВН+сопряженная кислота НАкислота Û Н+ + А-сопряженное основание Сопряженные кислотно-основные пары В /ВН+ и А- /НА называют буферными системами. Классификация кислотно-основных буферных систем. Буферные системы могут быть четырех типов: 1. Слабая кислота и ее анион А- /НА: · ацетатная буферная система СН3СОО-/СН3СООН в растворе СН3СООNa и СН3СООН, область действия рН 3, 8 – 5, 8. · Водород-карбонатная система НСО3-/Н2СО3 в растворе NaНСО3 и Н2СО3, область её действия – рН 5, 4 – 7, 4. 2. Слабое основание и его катион В/ВН+: · аммиачная буферная система NH3/NH4+ в растворе NH3 и NH4Cl, область ее действия – рН 8, 2 – 10, 2. 3. Анионы кислой и средней соли или двух кислых солей: · карбонатная буферная система СО32- /НСО3- в растворе Na2CO3 и NaHCO3, область ее действия рН 9, 3 – 11, 3. · фосфатная буферная система НРО42-/Н2РО4- в растворе Nа2НРО4 и NаН2РО4, область ее действия рН 6, 2 – 8, 2. Эти солевые буферные системы можно отнести к 1-му типу, т. к. одна из солей этих буферных систем выполняет функцию слабой кислоты. Так, в фосфатной буферной системе анион Н2РО4- является слабой кислотой. 4. Ионы и молекулы амфолитов. К ним относят аминокислотные и белковые буферные системы. Если аминокислоты или белки находятся в изоэлектрическом состоянии (суммарный заряд молекулы равен нулю), то растворы этих соединений не являются буферными. Они начинают проявлять буферное действие, когда к ним добавляют некоторое количество кислоты или щелочи. Тогда часть белка (аминокислоты) переходит из ИЭС в форму “белок-кислота” или соответственно в форму “белок-основание”. При этом образуется смесь двух форм белка: (R – макромолекулярный остаток белка) а) слабая “белок-кислота” + соль этой слабой кислоты: б) слабое “белок-основание” + соль этого слабого основания: Таким образом, и этот тип буферных систем может быть отнесен соответственно к буферным системам 1-го и 2-го типов.
Механизм буферного действия можно понять на примере ацетатной буферной системы СН3СОО-/СН3СООН, в основе действия которой лежит кислотно-основное равновесие: СН3СООН Û СН3СОО- + Н+; (рКа = 4, 8) Главный источник ацетат-ионов – сильный электролит СН3СООNa: СН3СООNa ® СН3СОО- + Na+ При добавлении сильной кислоты сопряженное основание СН3СОО- связывает добавочные ионы Н+, превращаясь в слабую уксусную кислоту: СН3СОО- + Н+ Û СН3СООН (кислотно-основное равновесие смещается влево, по Ле Шателье) Уменьшение концентрации анионов СН3СОО- точно уравновешивается повышение концентрации молекул СН3СООН. В результате происходит небольшое изменение в соотношении концентраций слабой кислоты и ее соли, а следовательно, и незначительно изменяется рН. При добавлении щелочи протоны уксусной кислоты (резервная кислотность) высвобождаются и нейтрализуются добавочные ионы ОН-, связывая их в молекулы воды: СН3СООН + ОН- Û СН3СОО- + Н2О (кислотно-основное равновесие смещается вправо, по Ле Шателье) В этом случае также происходит небольшое изменение в соотношении концентраций слабой кислоты и ее соли, а следовательно, и незначительное изменение рН. Уменьшение концентрации слабой кислоты СН3СООН точно уравновешивается повышение концентрации анионов СН3СОО- Способность буферного раствора сохранять рН по мере прибавления сильной кислоты или приблизительно на постоянном уровне далеко небеспредельна и ограничена величиной так называемой буферной емкости В. За единицу буферной емкости обычно принимают емкость такого буферного раствора, для изменения рН которого на единицу требуется введение сильной кислоты или щелочи в количестве 1 моль эквивалента на 1л раствора. Т. е. это величина, характеризующая способность буферного раствора противодействовать смещению реакции среды при добавлении сильных кислот или сильных оснований. Буферная емкость, как следует из ее определения, зависит от ряда факторов: 1. Чем больше количества компонентов кислотно-основной пары основание/ сопряженная кислота в растворе, тем выше буферная емкость этого раствора (следствие закона эквивалентов). 2. Буферная емкость зависит от соотношения концентраций компонентов буферного раствора, а следовательно, и от рН буферного раствора. главные буферные системы в организме человека Организм человека располагает тонкими механизмами координации происходящих в не физиологических и биохимических процессов и поддержания постоянства внутренней среды (оптимальных значений рН и уровней содержания различных веществ в жидкостях организма, температуры, кровяного давления и т. д.) Нередко отклонения рН крови от нормального для нее значения 7,36 всего лишь на несколько сотых приводят к неприятным последствиям. При отклонениях порядка 0,3 единицы в ту или другую сторону может наступит тяжелое коматозное состояние, а отклонения порядка 0,4 единицы могут повлечь даже смертельный исход. Впрочем, в некоторых случаях, при ослабленном иммунитете, для этого оказывается достаточными и отклонения порядка 0,1 единицы рН. Особенно большое значение буферных систем имеют в поддержании кислотно-основного равновесия организма. Внутриклеточные и внеклеточные жидкости всех живых организмов, как правило, характеризуются постоянным значением рН, которое поддерживается с помощью различных буферных систем. Значение рН большей части внутриклеточных жидкостей находится в интервале от 6,8 до 7,8. Кислотно-основное равновесие в крови человека обеспечивается водородкарбонатной, фосфатной и белковой буферными системами. Нормальное значение рН плазмы крови составляет 7,40 ± 0,05. Этому соответствует интервал значений активной кислотности а (Н+) от 3,7 до 4,0 ´ 10-8 моль/л. Так как в крови присутствуют различные электролиты - НСО3-, Н2СО3, НРО42-, Н2РО4-, белки, аминокислоты, это означает, что они диссоциируют в такой степени, чтобы активностьа (Н+) находилась в указанном интервале. 1. Водородкарбонатная (гидро-, бикарбонатная) буферная система НСО3-/Н2СО3 плазмы крови характеризуется равновесием молекул слабой угольной кислоты Н2СО3 с образующимися при ее диссоциации гидрокарбонат-ионами НСО3-(сопряженное основание): НСО3- + Н+ Û Н2СО3 НСО3- + Н2О Û Н2СО3 + ОН- В организме угольная кислота возникает в результате гидратации диоксида углерода – продукта окисления углеводов, белков и жиров. Причем процесс этот ускоряется под действием фермента карбоангидразы: СО2(р) + Н2О Û Н2СО3
2. Фосфатная буферная система НРО42-/Н2РО4- состоит из слабой кислоты Н2РО4-и сопряженного основания НРО42-. В основе ее действия лежит кислотно-основное равновесие, равновесие между гидрофофсфат- и дигидрофосфат-ионами: НРО42- + Н+ Û Н2РО4- , НРО42- + Н2О Û Н2РО4- + ОН- Фосфатная буферная система способа сопротивляться изменению рН в интервале 6, 2 – 8, 2, т. е. обеспечивает значительную долю буферной емкости крови. 3. Буферная система оксигемоглобин-гемоглобин, на долю которой приходится около 75% буферной емкости крови, характеризующаяся равновесием между ионами гемоглобина Hb- и самим гемоглобином HНb, являющимся очень слабой кислотой (КHНb = 6, 3 ´ 10-9; рКHНb = 8, 2). Hb- + Н+ Û HНb,,,Hb- + Н2О Û HНb + ОН- а также между ионами оксигемоглобина HbО2- и самим оксигемоглобином HНbО2, который является несколько более сильной, чем гемоглобин, кислотой (КHНbО2 = 1. 12 ´ 10-7; рКHНbО2 = 6, 95): HbО2- + Н+ Û HНbО2, HbО2- + Н2О Û HНbО2 + ОН-. Гемоглобин HНb, присоединяя кислород, образует оксигемоглобин HНbО2 , HНb + О2 Û HНbО2 и, таким образом, первые два равновесия взаимосвязаны со следующими двумя.
Вопрос 17Комплексными соединениями называются вещества молекулы которых состоят из центрального атома(или иона) М, непосредственно связанного с определенным числом n других молекул (или ионов) L., называемых лигандами.Комплексные соединения можно рассматривать как сложные соединения высшего порядка, состоящие из простых молекул способных к самостоятельному существованию в растворе.
По координационной теории Вернера в каждом комплексном соединении различают внутреннюю и внешнюю сферу. Центральный атом с окружающими его лигандами образуют внутреннюю сферу комплекса. Ее обычно заключают в квадратные скобки. Все остальное в комплексном соединении составляет внешнюю сферу и пишется за квадратными скобками. Общая запись формулы комплексного соединения имеет вид [MLn]Xm где М центральный атом,L-лиганд, Х-внешнесферная частица (молекула или ион) в квадратн скобки заключены частицы внутренней сферы. Центральный атом координирует лиганды, геометрически правильно располагая их в пространстве. Поэтому комплексные соединения частно назыв координационными . Число n лигандов соответственно координац числом, а внутренняя сфера координационной. Например [Сu(NH3)4]SO4/ Сu-центральный атом. Лиганлами явл. Молекулы аммиака. NH3 координац число n=4. Во внешнюю сферу уходит сульфат анион.SO42-Вокруг центрального атома размещается определенное число лигандов, которое определяется координационным числом (кч). Число координированных лигандов чаще всего равно 6 или 4. Лиганд занимает около центрального атома координационное место. При координации изменяются свойства как лигандов, так и центрального атома. Более прочно связанные частицы внутренней сферы называются комплексом (комплексным ионом). Между центральным атомом и лигандами действуют силы притяжения (образуется ковалентная связь по обменному и (или) донорно–акцепторному механизму), между лигандами – силы отталкивания. Если заряд внутренней сферы равен 0, то внешняя координационная сфера отсутствует.
Центральный атом (комплексообразователь) – атом или ион, который занимает центральное положение в комплексном соединении. Роль комплексообразователя чаще всего выполняют частицы, имеющие свободные орбитали и достаточно большой положительный заряд ядра, а следовательно могут быть акцепторами электронов. Это катионы переходных элементов. Наиболее сильные комплексообразователи – элементы IВ и VIIIВ групп. Редко в качестве комплексообразователей выступают нейтральные атомы d–элементов [Fe(CO)5] и атомы неметаллов в различной степени окисления [PF6]—. Число свободных атомных орбиталей, предоставляемых комплексообразователем, определяет его координационное число. Величина координационного числа зависит от многих факторов, но обычно она равна удвоенному заряду иона комплексообразователя: Лиганды – ионы или молекулы, которые непосредственно связаны с комплексообразователем и являются донорами электронных пар. Это электроноизбыточные системы, имеющие свободные и подвижные электронные пары, могут быть донорами электронов, например:
Cl— ¯; F—¯; ОН—¯; CN—¯; CNS—¯; Н2О¯; NH 3¯; СО¯.
Число мест, занимаемых каждым лигандом во внутренней сфере комплексного соединения называется координационной емкостью (дентатностью) лиганда. Она определяется числом электронных пар лиганда, которые участвуют в образовании координационной связи с центральным атомом.
Соединения р–элементов проявляют комплексообразующие свойства и выступают в комплексном соединении в качестве лигандов. Лигандами могут быть атомы и молекулы (белка, аминокислот, нуклеиновых кислот, углеводов). По числу связей, образуемых лигандами с комплексообразователем, лиганды делятся на моно-, би- и полидентатные лиганды. Вышеперечисленные лиганды – молекулы и анионы являются монодентатными, так как они доноры одной электронной пары. К бидентатным лигандам относятся молекулы или ионы, содержащие две функциональные группы, способные быть донором двух электронных пар( – ООС — СОО –). К полидентатные лигандам можно отнести 6–дентатный лиганд этилендиаминтетрауксусной кислоты.
18. Пространственное строение комплексных соединений. Классы комплексных соединений.Пространственное строение комплексаопределяется типом гибридизации атомных орбиталей центрального атома. Комплексные соединения с координационным числом 2 встречаются редко(sp-гибридизация).пример:[Ag(NH3)2]+. Наиболее распространены комплексы с координационными числами 4 и 6. Для описания геометрии комплексных соединений пользуются понятием координационных полиэдров(многогранник.вершинами которого служат лиганды, связанные с центральным атомом-комплексообразователем. Комплексы с координационным числом 4 могут быть как тетраэдрическими(sp3-гибридизация)пример:[Zn(NH3)4]2+ , так и плоскоквадратными(dsp2-гибридизация)пример:[Pt(NH3)4]2+. Комплексы с координационным числом 6 имеют октаэдрическую конфигурацию(d2sp3-гибридизация).Пример:[Ni(NH3)6]3+. Реже встречаются комплексы с координационным числом 5(dsp3-гибридизация). Они образуют тригональную бипирамиду(пример:[CdCI5]3-) или квадратную пирамиду.( пример:[Ni(CN)5]3-). Координационному числу 12(очень редко) соответствует икосаэдр.
Классы комплексных соединений: 1)Катионные комплексыобразованы в результате координации вокругположительного ионанейтральных молекул (H2O, NH3и др.). [(Zn(NH3)4)]Cl2— хлорид тетраамминцинка(II) [Co(NH3)6]Cl3— хлорид гексоамминкобальта(II) 2)Анионные комплексы: в роли комплексообразователя выступает атом с положительнойстепенью окисления, а лигандами являются простые или сложные анионы. K2[BeF4] — тетрафторобериллат(II) калия Li[AlH4] —тетрагидридоалюминат(III) литияK3[Fe(CN)6] —гексацианоферрат(III) калия
3) Нейтральные комплексыобразуются при координации молекул вокруг нейтрального атома, а так же при одновременной координации вокругположительного иона— комплексообразователяотрицательных ионовимолекул. [Ni(CO)4] — тетракарбонилникель [Pt(NH3)2Cl2] — дихлородиамминплатина(II)
4)Внутрикомплексные клешневидные соединения, хелатные соединения. Классический пример— гликоколят меди
\
19. Комплексоны, их применение в медицине. Ионные равновесия в растворах комплексных соединений. Константа нестойкости комплексного иона. Сложные органические лиганды. Представление о строении металлоферментов. Механизм токсического действия тяжёлых металлов на основе ЖМКО.
Комплексоны -аминополикарбоновые кислоты и их производные, применяемые в методе комплексонометрии, а также для умягчения воды и др. технических целей. В аналитической практике используют нитрилотриуксусную кислоту N (CH2COOH)3 — комплексон I и двунатриевую соль этой кислоты — комплексон III, илитрилон Б.
Вещества, устраняющие последствия воздействия ядов на биологические структуры и инактивирующие яды, посредством химических реакций, называют антидотами.
(Na3 СаДТПА) – пентацин и (NaCa2 ДТПФ) –тримефацин. Их применяют при острых и хроническихотравлениях свинцом, радионуклидами, алюминием, цинком, цериеми др, (Na2СаЭДТФ)фосфицинуспешно используется для выведения из организмартути, свинца, берилия, марганца, актиноидов и других металлов.
В водных растворах комплексных соединений устанавливается равновесие, которое характеризуется константой устойчивости (Куст.) или величиной, обратной ей, константой нестойкости (Кн). Пользуясь величиной соответствующей константы, необходимо уметь рассчитывать равновесные концентрации ионов в растворе комплексных соединений в присутствии избытка лиганда и без избытка лиганда.
константа
равновесия - константа нестойкости
комплексного иона [Ag(NH3)2]+ :
![]() Наиболее устойчивые комплексные
соединения имеют наименьшие константы
нестойкости.
Наиболее устойчивые комплексные
соединения имеют наименьшие константы
нестойкости.
У сложных белков, кроме белковой цепи, имеется дополнительная небелковая группа. Она называется лиганд, то есть молекула, связанная с белком.
В роли лиганда могут выступать любые молекулы:
молекулы, выполняющие в белке структурную функцию – липиды, углеводы, нуклеиновые кислоты, минеральные элементы, какие-либо другие органические соединения: гем в гемоглобине, углеводы в гликопротеинах, ДНК и РНК в нуклеопротеинах, медь в церулоплазмине,
переносимые белками молекулы: железо в трансферрине, гемоглобин в гаптоглобине, гем в гемопексине,
субстраты для ферментов – любые молекулы и даже другие белки.
Металлоферменты— ферментоы, для функционирования которых необходимо присутствие катионов тех или иных металлов. В подобном ферменте могут присутствовать несколько различных ионов металла. Катион металла при этом обеспечивает правильную пространственную конфигурацию активного центра металлофермента.
Примерами металлоферментов являются карбоксипептидаза,карбоангидраза, илиселен-зависимая монодейодиназа,конвертирующая тироксин в трийодтиронин
ЖМКО
Мягкие кислоты предпочтительней координируют мягкие основания, а жёсткие – жёсткие. Таким образом, ионы ртути, свинца, таллия (мягкие кислоты) имеют большее сродство к серосодержащим лигандам, а ионы магния, кальция (жёсткие) – к кислород- и азотсодержащим лигандам.
20) Понятие биогенности химических элементов. Биосфера, круговорот биогенных элементов. Классификация биогенных элементов по их функциональной роли: органогены, элементы электролитного фона, микроэлементы, ксенобиотики.
Элементы необходимые организму для построения и жизнедеятельности клеток и органов, называют биогенными элементами.
Часть земной оболочки, занятой растительными и животными организмами, переработанная ими и космическими излучениями, приспособленная к жизни, называют биосферой.
Концентрация элементов в живом веществе прямо пропорциональна его содержанию в среде обитания с учетом растворимости их соединений. Химический состав организма определяется составом окружающей среды. Биосфера содержит 100 млрд. тонн живого вещества. Около 50% массы земной коры приходится на кислород, более 25% на кремний. Восемнадцать элементов (О, Si, Al, Fe, Ca. Na, К, Mg, H, Ti, С, Р, N, S, Cl, F, Мn, Ва) составляют 99,8% массы земной коры. Живые организмы принимают активное участие в перераспределении химических элементов в земной коре. Минералы, природные химические вещества, образуются в биосфере в различных количествах, благодаря деятельности живых веществ (образование железных руд, горных пород, в основе которых соединения кальция). Кроме этого, оказывают влияние техногенные загрязнения окружающей среды. Изменения, происходящие в верхних слоях земной коры, влияют на химический состав живых организмов. В организме можно обнаружить почти все элементы, которые есть в земной коре и морской воде. Пути поступления элементов в организм разнообразны. Согласно биогеохимической теории Вернадского существует «биогенная миграция атомов» по цепочке воздух> почва>вода>пища>человек, в результате которой ,практически все элементы, окружающие человека во внешней среде, в большей или меньшей степени проникают внутрь организма.
Для 30 элементов биогенность установлена.
Классификация биогенных элементов по их функциональной роли:
1) органогены, в организме их 97,4% (С, Н, О, N, Р, S),
2) элементы электролитного фона (Na, К, Ca, Mg, Сl). Данные ионы металлов составляют 99% общего содержания металлов в организме;
3) Микроэлементы – это биологически активные атомы центров ферментов, гормонов (переходные металлы).
Ксенобиотики - чужеродные для организмов химические вещества, естественно не входящие в биотический круговорот и прямо или косвенно порожденные хозяйственной деятельностью человека.
21. Квантово-механическая модель атома. Применение системы квантовых чисел для характеристики энергетического состояния электрона. Основное и возбужденное состояние атома. В основу КММ положена квантовая теория атома, согласно которой электрон обладает как свойствами частицы, так и свойствами волны (корпускулярно-волновой дуализм). Другими словами, о местоположении электрона в определенной точке можно судить не точно, а с определенной долей вероятности. Поэтому в КММорбитыБора заменилиорбиталями(область пространства внутри атома, в которой сосредоточена большая часть заряда электрона – совокупность вероятных положений электрона). Состояние электрона в атоме описывают с помощью 4 чисел, которые называют квантовыми (определены с помощью волнового уравнения Шредингера):
|
Квантовое число |
Символ |
Описание |
Значения |
|
Главное |
n |
Энергетический уровень орбитали |
Положительные целые числа: 1, 2, 3... |
|
Орбитальное (побочное) |
L |
Форма орбитали |
Целые числа от 0 до n-1 |
|
Магнитное |
m |
Ориентация |
Целые числа от -Lдо +L |
|
Спиновое |
s |
Спин электрона |
+½ и -½ |
Главное квантовое число n. Описывает среднее расстояние от орбитали до ядра и энергетическое состояние электрона в атоме. Чем больше значение n, тем выше энергия электрона и больше размер электронного облака. Если в атоме несколько электронов с одинаковым n, то они образуют электронные облака одинакового размера – энергетические уровни. Максимально возможное значенияnдля электронов невозбужденного атома данного элемента соответствует номеру периода, в котором находится этот элемент. Чем больше значениеn, тем большее число электронов может находиться на данном энергетическом уровне. Емкость энергетического уровня вычисляют по формуле 2n2. При значенияхn>1 наблюдается расщепление энергетического уровня на подуровни. Это означает, что электроны, находящиеся на одном энерг уровне несколько различаются по запасу энергии и формами атомных орбиталей. Число подуровней на данном энергетическом уровне равно главному квантовому числу этого уровня. Орбитальный радиус rор– расстояние от ядра атома до наиболее удаленной точки поверхности, изображающей атомную орбиталь.Орбитальное квантовое число lОписывает форму орбитали, которая зависит от n.Орбитальное число l может принимать целочисленные значения в диапазоне от 0 до n-1. Например, при n=2: l=0 l=1 Значение l определяет форму орбитали, а n – ее размер. Орбитали, имеющие одинаковое n, но разные l называютэнергетическими подуровнямии обозначают буквами латинского алфавита:
|
l |
Энергетический подуровень |
|
0 1 2 3 4 |
S (сферическая форма) p (гантелеобразная) d (клеверный лист) f g |
Магнитное квантовое число m. Описывает ориентацию орбиталей в пространстве.Может принимать целочисленные значения в диапазоне от -l до +l (включая 0). Например: Для l=0 возможно только одно значение: m=0. Это значит, что s-орбиталь имеет только одну пространственную ориентацию. Для l=1: m=-1;0;+1 - p-орбиталь имеет три пространственные ориентации. Для l=2: m=-2;-1;0;+1;+2 - d-орбиталь имеет пять пространственных ориентаций. Три квантовые числа характеризуют атомные орбитали, четвертое квантовое число – только электрон.Спиновое квантовое число s. Описывает направление вращения электрона в магнитном поле - по часовой стрелке или против. На каждой орбитали может находиться только два электрона: один со спином +½ другой -½. При изображении электронных конфигураций электроны, имеющие противоположный спин, обозначаются противоположно направленными стрелками. Орбиталь и соответствующий энергетический уровень, определяемый четырьмя квантовыми числами n, l, m, s называют квантовым состоянием электрона в атоме. Основное и возбужденное состояние атома. Состояние атома с минимально возможной энергией электронов в нем называется основным или невозбужденным состоянием. В этом состоянии атома все электроны в нем подчиняются принципу минимальной энергии (занимают в первую очередь орбитали, имеющие наименьшую энергию). При получении энергии извне, один либо несколько электронов могут повышать свою энергию, поднимаясь на более высокие энергетические состояния. Такие состояния атома называются возбужденными. Возбужденное состояние характеризуется избыточной, по сравнению с основным, энергией. Переход атома в возбужденное состояние происходит при облучении или нагревании вещества. Состояние которое требует для повышения энергии электрона наименьших энергетически затрат называют первым возбужденным состоянием. Возбужденное состояние атома является неустойчивым и через некоторое время (8-10 секунд) электрон теряет энергию, перейдя на энергетическую орбиталь с меньшей энергией, испустив при этом квант света. Так, к примеру, электронную конфигурацию атома гелия в первом возбужденном состоянии можно записать так 1s12s1
22Ковалентная
связь (атомная
связь, гомеополярная связь) — химическая
связь,
образованная перекрытием (обобществлением)
пары валентных электронных
облаков.
Обеспечивающие связь электронные облака
(электроны) называются общей
электронной парой.
Характерные
свойства ковалентной связи —
направленность, насыщаемость, полярность,
поляризуемость — определяют химические
и физические свойства соединений.
Направленность связи обусловлена
молекулярным строением вещества и
геометрической формы их молекулы. Углы
между двумя связями называют валентными.
Насыщаемость — способность атомов
образовывать ограниченное число
ковалентных связей. Количество связей,
образуемых атомом, ограничено числом
его внешних атомных орбиталей. Полярность
связи обусловлена неравномерным
распределением электронной плотности
вследствие различий в электроотрицательностях
атомов. По этому признаку ковалентные
связи подразделяются на неполярные и
полярные. Поляризуемость связи выражается
в смещении электронов связи под влиянием
внешнего электрического поля, в том
числе и другой реагирующей частицы.
Поляризуемость определяется
подвижностью электронов.
Полярность и поляризуемость ковалентных
связей определяет реакционную способность
молекул по отношению к полярным реагентам.
Электроны тем подвижнее, чем дальше они
находятся от ядер. Ковалентная связь
это когда два атома делятся электронами
и держатся вместе.а)
Ковалентная неполярная
связь образуется между атомами неметалла
одного и того лее химического элемента.
Такую связь имеют простые вещества,
например О2; N2; C12. Можно привести схему
образования молекулы водорода: ![]() (на
схеме электроны обозначены точками).
б)
Ковалентная полярная
связь образуется между атомами различных
неметаллов. Схематично образование
ковалентной полярной связи в молекуле
НС1 можно изобразить так:
(на
схеме электроны обозначены точками).
б)
Ковалентная полярная
связь образуется между атомами различных
неметаллов. Схематично образование
ковалентной полярной связи в молекуле
НС1 можно изобразить так:
![]() Общая
электронная плотность оказывается
смещенной в сторону хлора, в результате
чего на атоме хлора возникает частичный
отрицательный заряд
Общая
электронная плотность оказывается
смещенной в сторону хлора, в результате
чего на атоме хлора возникает частичный
отрицательный заряд ![]() ,
а на атоме водорода — частичный
положительный
,
а на атоме водорода — частичный
положительный ![]() .
Таким образом, молекула становится
полярной:
.
Таким образом, молекула становится
полярной: ![]() Образование
связи по донорно-акцепторному механизму
существует
гетерогенный механизм — взаимодействие
разноименно заряженных ионов — протона H+ и
отрицательного иона водорода H-,
называемого гидрид-ионом:H+ +
H- →
H2
При
сближении ионов двухэлектронное облако
(электронная пара) гидрид-иона притягивается
к протону и в конечном счёте становится
общим для обоих ядер водорода, то есть
превращается в связывающую электронную
пару. Частица, поставляющая электронную
пару, называется донором, а частица,
принимающая эту электронную пару,
называется акцептором. Такой механизм
образования ковалентной связи называется
донорно-акцепторным.[7]
Распределение электронной
плотности между
ядрами в молекуле водорода одно и то
же, независимо от механизма образования,
поэтому называть химическую связь,
полученную по донорно-акцепторному
механизму, донорно-акцепторной связью
не корректно.
В
качестве донора электронной пары, кроме
гидрид-иона, выступают соединения
элементов главных подгрупп V—VII
групп периодической
системы элементов в
низшей степени окисления элемента. Так,
ещё Йоханнес
Брёнстед установил,
что протон не существует в растворе в
свободном виде, в воде он образует
катион оксония:
H+ +
H2O
→ H3O+
Образование
связи по донорно-акцепторному механизму
существует
гетерогенный механизм — взаимодействие
разноименно заряженных ионов — протона H+ и
отрицательного иона водорода H-,
называемого гидрид-ионом:H+ +
H- →
H2
При
сближении ионов двухэлектронное облако
(электронная пара) гидрид-иона притягивается
к протону и в конечном счёте становится
общим для обоих ядер водорода, то есть
превращается в связывающую электронную
пару. Частица, поставляющая электронную
пару, называется донором, а частица,
принимающая эту электронную пару,
называется акцептором. Такой механизм
образования ковалентной связи называется
донорно-акцепторным.[7]
Распределение электронной
плотности между
ядрами в молекуле водорода одно и то
же, независимо от механизма образования,
поэтому называть химическую связь,
полученную по донорно-акцепторному
механизму, донорно-акцепторной связью
не корректно.
В
качестве донора электронной пары, кроме
гидрид-иона, выступают соединения
элементов главных подгрупп V—VII
групп периодической
системы элементов в
низшей степени окисления элемента. Так,
ещё Йоханнес
Брёнстед установил,
что протон не существует в растворе в
свободном виде, в воде он образует
катион оксония:
H+ +
H2O
→ H3O+
Протон атакует неподелённую электронную пару молекулы воды и образует устойчивый катион, существующий в водных растворах кислот.[8]Аналогично происходит присоединение протона к молекуле аммиака с образованием комплексного катиона аммония:NH3 + H+ → NH4+ Таким путём (по донорно-акцепторному механизму образования ковалентной связи) получают большой класс ониевых соединений, в состав которого входят аммониевые, оксониевые, фосфониевые, сульфониевые и другие соединения В качестве донора электронной пары может выступать молекула водорода, которая при контакте с протоном приводит к образованию молекулярного иона водорода H3+: H2 + H+ → H3+ Связывающая электронная пара молекулярного иона водорода H3+ принадлежит одновременно трём протонам.
23.
Химия элементов s
блока. К
s-элементам относятся две группы
Периодической системы: IА и IIА. В группу
IА входят 8 элементов: литий, калий,
натрий, рубидий, цезий, франций, водород,
гелий. В группу IIА входят 6 элементов:
бериллий, магний, кальций, стронций,
барий, радий. Общая характеристика
элементов IА и IIА. Элементные вещества
- типичные металлы, обладающие блеском,
высокой электрической проводимостью
и теплоповодимостью, химически весьма
активны.s-элементы IА и IIА имеют относительно
большие радиусы атомов и ионов. s-элементы
IА и IIА групп легко отдают валентные
электроны. Являются сильными
восстановителями. С ростом радиуса
атома в группах IА и IIА ослабевает связь
валентных электронов с ядром, следовательно
s-элементы этих групп имеют низкие
значения Еи и Еср..сильных восстановителей.
Восстановительные свойства возрастают
закономерно с увеличением радиуса
атома. Восстановительная способность
увеличивается по группе сверху вниз.
Для элементов IIА группы характерна
большая, чем для элементов IА группы
способность к комплексообразованию.
s-элементы IА и IIА образуют соединения
с ионным типом связи. Исключение
составляет водород, для которого в
соединениях даже с самыми электроотрицательными
элементами характерна преимущественно
ковалентная связь (например, фтороводород
или вода.Сравнение
свойств элементов IА и IIА (комплексообразование,
образование осадков) на примере Na, K и
Mg, Ca Атомы
элементов IА группы имеют по одному
валентному электрону на s подуровне
внешнего энергетического уровня. Это
обуславливает проявление степени
окисления +1![]()
Химическая активность металлов IА группы возрастает закономерно с увеличением радиуса атома и уменьшением их способности к гидратированию (чем меньше способность к гидратированию, тем активнее металл). Так как радиус атома калия больше, чем радиус атома натрия, то способность к гидратации для катиона калия будет ниже, чем для катиона натрия, а, следовательно, химическая активность катиона калия выше, чем у катиона натрия. Вследствие незначительного поляризующего действия (устойчивая электронная структура, большие размеры, малый заряд ядра) комплексообразование для ионов щелочных металлов малохарактерно. Вместе с тем, они способны образовывать комплексные соединения с некоторыми биолигандами (КЧ для натрия и калия может принимать значения 4 и 6). Способность образовывать донорно-акцепторные связи с соответствующими лигандами едва намечается у натрия. У калия имеется значительная тенденция к использованию имеющихся в атоме вакантных d-орбиталей. Большинство солей щелочных металлов хорошо растворимы в воде (исключение составляют некоторые соли лития). Степени окисления больше +2 элементы IIА группы не проявляют. Несмотря на то, что число валентных s электронов у атомов IIА группы одинаково, свойства магния и кальция отличаются друг от друга. Это связанно с тем, что в атоме кальция, в отличие от атома магния, имеются свободные d-орбитали, близкие по энергии к ns орбиталям. Ионы натрия играют важную роль в обеспечении постоянства внутренней среды человеческого организма, участвуют в поддержании постоянного осмотического давления биожидкости (осмотического гомеостаза) Ионы натрия участвуют в регуляции водного обмена и влияют на работу ферментов.. Ионы калия играют важную роль в физиологических процессах - сокращении мышц, нормальном функционировании сердца, проведении нервных импульсов, обменных реакциях. Являются важными активаторами внутриклеточных ферментов. Формально магний относится к макроэлементам. В наибольшей степени магний концентрируется в дентине и эмали зубов, костной ткани. Накапливается в поджелудочной железе,скелетных мышцах. Ионы кальция принимают активное участие в передаче нервных импульсов, сокращении мышц, регулировании работы сердечной мышцы, механизмах свертывания крови. Химическое сходство и биологический антагонизм натрия, калия, кальция и магния.Сходство электронного строения ионов щелочных (натрий и калий) и щелочноземельных (магний и кальций) металлов и различия физико-химических характеристик определяет их действия на биологические процессы.Натрий и калий являются антагонистами. В ряде случаев близость многих физико-химических свойств обусловливает их взаимозамещение в живых организмах. Например, при увеличении количества натрия в организме усиливается выведение калия почками, наступает гипокалиемия.Магний и кальций являются антагонистами. Ионы кальция подавляют активность многих ферментов, активизируемых ионами магния. Антагонизм ионов кальция и магния проявляется еще и в том, что ион кальция является внеклеточным ионом. При длительном поступлении в организм избыточных количеств солей магния наблюдается усиленное выделение кальция из костной ткани.
24.химия элементов d-блока.Электронные структуры атомов и катионов.Наиболее важные элементы.Биологическая роль железа,марганца,хрома,цинка,серебра,меди.Образование комплексных соединений.Гидроксокомплексы;амминокомплексы;образование нерастворимых соединений;Аналитические реакции на некоторые катионы д-элементов.
Они расположены в побочных подгруппах,между s- и p-элементами. (ScTiVCrMnFeCoCuZnи т.д) Все они металлические элементы.Характерная особенность: в их атомах последними заполняются орбитали не внешнего слоя (как у s- и p-элементов), а предвнешнего [(n - 1)d] слоя,поэтому в пределах одного периода свойства меняются не резко. Немонотонность(наполовину заполненный или полностью заполненный подуровень является устойчивой конфигурацией).Свойства: t плавления и плотность монотонно возрастают с увеличение порядкового номера,атомные радиусы возрастают,энергия ионизации наоборот уменьшается. Выделяют семейство железа(Fe,Co,Ni) семейство платиновых металлов(Ru,Rh,Pd,Os,Ir,Pt)в связи в эффектом f-сжатия-химические свойства схожи,т.к их атомные радиусы практически одинаковые.Металлические свойства убывают в периодах слева направо,а в группах-сверху вниз. Элементы d-блока находящиеся в III, IV, V, VI, VII B группах имеют незавершенный d-электронный слой (предвнешний эн. уровень).Такие электронные оболочки неустойчивы.Этим объясняется переменная валентность и возможность проявлять различные степени окисления d-элементов. Степени окисления элементов d-блока в соединениях всегда только положительные. Соединения с высшей степенью окисления проявляют кислотные и окислительные свойства (в растворах представлены кислородсодержащими анионами).Соединения с низшей степенью окисления – основные и восстановительные свойства (в растворах представлены катионами). Соединения с промежуточной степенью окисления – проявляют амфотерные свойства.Например: CrOосновной оксид, Cr2O3– амфотерный оксид, CrO3– кислотный оксид.
Fe1s22s22p63s23p64s23d6 Cr1s22s22p63s23p64s23d54s1 - «Провал электрона» Важные элементы:Fe,Mn,Zn,Cu,Ag,PtFe - ходит в состав гемоглобина, ферментов цитохромов, каталазы, пероксидазы Co - входит в состав витамина В12 Cr - Биогенный элемент. Mn - Входит в состав ферментов. Cu - Входит в состав ферментов окигеназ и гидролаз. Участвует в кроветворении. Zn - Входит в состав ферментов катализирующих гидролиз пептидов, белков, некоторых эфиров и альдегидов. Комплексообразующая способность d-элементов Возможность создания химических связей с участием d-электронов и свободных d-орбиталей обуславливает ярко выраженную способность d-элементов к образованию устойчивых комплексных соединений. При низких степенях окисления для d-элементов более характерны катионные, а при высоких – анионные октаэдрические комплексы ([ScF6]3–, [TiF6]2–, [VF6]–) КЧисла d-элементов непостоянны, это четные числа от 4 до 8, реже 10,12. Используя незаполненные d-орбитали и неподеленные пары d-электронов на предвнешнем электронном слое, d-элементы способны выступать как донорами электронов –дативная связь, так и акцепторами электронов. Пример соединений с дативной связью: [HgI]Ї, [CdCl4]Ї. Гидроксокомплексы – комплексные соединения, содержащие в качестве лигандов гидроксид-ионы OH-. Гидроксокомплексы образуются в реакциях протолиза из аквакомплексов: [Al(H2O)6]3++ H2O =[Al(H2O)5(OH)]2+ + H3O+либо при растворении амфотерных гидроксидов в водных растворах гидроксидов щелочных металлов:Zn(OH)2+ 2OH- = [Zn(OH)4]2-Амминокомплекс(аммиакат)-комплексные соединения,где лиганды Nh3+ например:[Zn( NH3)4 ]2+ тетраэдрическая конфигурация sp3-гибридизацияАналитические реакции на Cu2+ Ag+ 2CuSO4+2NH3+2H20=(CUOH)2SO4 осадок+(NH4)2SO4(CuOH)2SO4+NH3(изб.)=Cu(NH3)4(OH)2синего цвета Ag+ +Cl2=AgCl (белый творожистый осадок)затем к нему добавляют избыток аммиака,в результате осадок переходит в неокрашенный раствор
23. К р – блоку относятся 30 элементов IIIA-VIIIA групп периодической системыи входят во второй и третий малые периоды, а также в четвертый – шестой большие периоды. У элементов IIIA группы появляется первый электрон на р – орбитали. В других группах IVA-VIIIA происходит последовательное заполнение р – подуровня до 6 электронов. Строение внешних электронных оболочек атомов элементов р – блока ns2npa , где а = 1÷6.
На свойства р–элементов и их соединений оказывает влияние как появление новых подуровней на внешней электронной оболочке, так и заполнение внутренних электронных оболочек. р – Элементы второго периода (В, С, N, O, F) резко отличаются от элементов нижеследующих периодов, так как, начиная с р–элементов третьего периода, появляется низколежащий свободный d-подуровень, на который могут переходить электроны с р – подуровня при возбуждении атома. Полностью заполненный 3 d-подуровень у р–элементов четвертого периода (Ga, Ge, As, Se, Br) обуславливает отличие их свойств от элементов третьего периода. Максимальное заполнение 4f-подуровня в шестом периоде сказывается на различии свойств р–элементов шестого и пятого периодов.
Вдоль периода у р–элементов падает способность к образованию положительно заряженных ионов с зарядом, отвечающим номеру группы, и наоборот, способность к образованию отрицательных ионов с зарядом, равным (8 – № группы) возрастает.
р – элементы образуют двухатомные молекулы Э2, различающиеся по устойчивости. Наиболее устойчивы молекулы элементов второго периода (N2, O2, F2). При переходе от IIIA к IVA и VA группам устойчивость двухатомных молекул возрастает, а затем при переходе к VIIIА группе понижается. В группах сверху вниз прочность связи Э–Э уменьшается.
Постоянные пломбировочные (реставрационные) материалы предназначены для восстановления анатомической формы, функции и внешнего вида зуба, а также предотвращения развития кариеса. Один из разновидностей таких материалов цементы:
– цинк-фосфатные цементы;
– силикатные цементы;
– силикофосфатные цементы.
Твердокристаллические материалы
К этой группе слепочных материалов относятся гипс, цинкоксиэвгеноловые и цинкоксигваяколовые пасты. Характерной особенностью этих масс является то, что в отвердевшем состоянии они имеют четкое кристаллическое строение, лишены пластичности и упругих свойств.
26.
Адсорбционные равновесия и процессы
на подвижных границах раздела фаз.
Поверхностное натяжение. Адсорбция.
Поверхностноактивные, неактивные и
инактивные вещества. Ориентация молекул
в поверхностном слое и структура
биомембран. Подвижные поверхности
раздела между жидкостью и газом (ж – г)
и двумя несмешивающимися жидкостями
(ж – ж). для таких систем справедливы
следующие понятия. Поверхностные
явления – процессы, происходящие на
границе раздела фаз и обусловленные
особенностями состава и строения
поверхностного (пограничного) слоя.
Поверхностная энергия Гиббса.
Gs = ơ * s,
гдеGs– поверхностная
энергия Гиббса системы, Дж; ơ –
коэффициент пропорциональности,
называемый поверхностным натяжением,
Дж/м2; s – межфазная поверхность,
м2. Поверхностное натяжение (ơ)- величина, численно равная энергии
Гиббса, приходящейся на единицу площади
поверхностного слоя и численно равная
работе, которую необходимо совершить
для образования единицы поверхности
раздела фаз при постоянной температуре. –Стремление в-ва (жидкости или
твердой фазы) уменьшить избыток своей
потенциальной энергии на границе раздела
с др. фазой (поверхностную энергию).
ПН зависит от природы жидкости и
температуры (уменьшается с ростомt).
Вода имеет самое высокое значение ПН.
Поверхностная активность (g)–способность растворенных веществ
изменять поверхностное натяжение
растворителя. [Дж] G=
- Адсорбция (Г) – самопроизвольное
изменение концентрации растворенного
вещества на границе раздела фаз.
[моль/см2; моль/м2].
Адсорбция (Г) – самопроизвольное
изменение концентрации растворенного
вещества на границе раздела фаз.
[моль/см2; моль/м2].
![]() ,
гдеR– универсальная
газовая постоянная (8,31 Дж/моль * К), с –
концентрация раствора (моль/л).
Адсорбентом называется
вещество, на поверхности которого
происходит изменение концентрации
другого вещества –адсорбата.
Происходит на любых межфазовых
поверхностях и адсорбироваться могут
любые вещества.Адсорбционное
равновесие, т.е. равновесие распределения
вещества между пограничным слоем и
граничащими фазами,является динамическим
равновесиеми быстро устанавливаются.
Адсорбция уменьшается с увеличением
температуры и покидает поверхность.
Зависимость адсорбции от равновесной
концентрации растворенного вещества
при постоянно температуре называютизотермой адсорбции.
,
гдеR– универсальная
газовая постоянная (8,31 Дж/моль * К), с –
концентрация раствора (моль/л).
Адсорбентом называется
вещество, на поверхности которого
происходит изменение концентрации
другого вещества –адсорбата.
Происходит на любых межфазовых
поверхностях и адсорбироваться могут
любые вещества.Адсорбционное
равновесие, т.е. равновесие распределения
вещества между пограничным слоем и
граничащими фазами,является динамическим
равновесиеми быстро устанавливаются.
Адсорбция уменьшается с увеличением
температуры и покидает поверхность.
Зависимость адсорбции от равновесной
концентрации растворенного вещества
при постоянно температуре называютизотермой адсорбции.
![]() изотерма
адсорбции ПАВ на границе раздела раствор
- газ
изотерма
адсорбции ПАВ на границе раздела раствор
- газ
Классификация веществ по влиянию на поверхностное натяжение растворителя1. Поверхностно-активные вещества (ПАВ) – понижают поверхностное натяжение растворителя g > 0, Г>0 (по отношению к воде – органические соединения дифильного строения). 2. Поверхностно-инактивные вещества (ПИВ) – незначительно повышают поверхностное натяжение растворителя g < 0, (неорганические кислоты, основания, соли, глицерин, и др). 3. Поверхностно-неактивные вещества (ПНВ) – практически не изменяют поверхностного натяжения растворителя g = 0, (по отношению к воде веществами являются сахароза и ряд других).
Ориентация молекул в поверхностном слое и структура биомембран. Поскольку только полярная головка ПАВ растворима в воде, а гидрофобный хвост нет, то такие молекулы выталкиваются на поверхность, где и накапливаются - это примерположительной адсорбции. Пределом такой адсорбции служит полное насыщение поверхностного слоя адсорбируемыми веществами. Если вещество увеличивает поверхностное натяжение, то оно втягивается во внутренние слои жидкости. Этоотрицательная адсорбция, поскольку поверхностные слои обедняются растворенным веществом. Лэнгмюр выдвинул предположение, что при максимальных величинах сорбции
поверхность раствора покрыта слоем толщиной в одну молекулу ПАВ (мономолекулярным
слоем), гидрофобные хвосты молекул ПАВ направлены перпендикулярно поверхности, образуя
подобие частокола. При вертикальной ориентации длина цепи молекулы не влияет на
площадь, занятую молекулой в поверхностном слое.
![]()
Структура плазматической мембраны Все биомембраны построены одинаково; они состоят из двух слоев липидных молекул толщиной около 6 нм, в которые встроены белки. Некоторые мембраны содержат, кроме того, углеводы, связанные с липидами и белками. Соотношение липиды : белки : углеводы является характерным для клетки или мембраны и существенно варьирует в зависимости от типа клеток или мембран. Компоненты мембран удерживаются нековалентными связями, вследствие чего они обладают лишь относительной подвижностью, т. е. могут диффундировать в пределах липидного бислоя. Текучесть мембран зависит от липидного состава и температуры окружающей среды. С увеличением содержания ненасыщенных жирных кислот текучесть возрастает, так как наличие двойных связей способствует нарушению полукристаллической мембранной структуры. Подвижными являются и мембранные белки. Если белки не закреплены в мембране, они «плавают» в липидном бислое как в жидкости. Поэтому говорят, что биомембраны имеют жидкостно-мозаичную структуру. В то время как «дрейф» в плоскости мембраны происходит достаточно легко, переход белков с внешней стороны мембраны на внутреннюю невозможен, а переход липидов происходит крайне редко. Для «перескока» липидов необходимы специальные белки транслокаторы. Исключение составляет холестерин, который может легко переходить с одной стороны мембраны на другую. Мембранные липиды На рисунке схематически изображена биомембрана. В мембранах содержатся липиды трех классов: фосфолипиды, холестерин и гликолипиды. Холестерин присутствует во внутриклеточных мембранах животных клеток (за исключением внутренней мембраны митохондрий). Гликолипиды входят в состав многих мембран (например, во внешний слой плазматических мембран). В состав гликолипидов входят углеводные функциональные группы, которые ориентируются в водную фазу.
![]()
27. Адсорбционные равновесия на неподвижных границах раздела фаз. Физическая адсорбция и хемосорбция. А. газов на твердых телах, а. из растворов, избирательная а., зависимость величины а. от различных факторов.
Адсорбция на неподвижных поверхностях раздела твердое-жидкое, газ-жидкость, где удельная поверхность энергии гиббса поддается экспериментальному определения работает уравнение Фрейндлиха: a=k*c (в степени n) Если адсорбция на твердом теле происходит из раствора, то нужно учитывать,что кроме молекул растворенного вещества могут адсорбироваться и молекулы растворителя. Физическая адсорбция возникает за счет ван-дер-ваальсовых взаимодействий. Она характеризуется обратимостью и уменьшением адсорбции при повышении температуры, т.е. экзотермичностью, причем тепловой эффект физической адсорбции обычно близок к теплоте сжижения адсорбата (10 – 80 кДж/моль). Таковой является, например, адсорбция инертных газов на угле. Физическая адсорбция является обратимым процессом, условие равновесия определяется равными скоростями адсорбции молекул адсорбтива P на вакантных местах поверхности адсорбента S* и десорбции — освобождения адсорбата из связанного состояния S − P:
![]() ; уравнение
равновесияя в таком случае:
; уравнение
равновесияя в таком случае:
![]() ,
где K —константа
равновесия, [S − P] и [S*] —
доли поверхности адсорбента, занятые
и незанятые адсорбатом, а [P] —
концентрация адсорбтива.Химическая
адсорбция (хемосорбция)
осуществляется путем химического
взаимодействия молекул адсорбента и
адсорбата. Хемосорбция обычно необратима;
химическая адсорбция, в отличие от
физической, является локализованной,
т.е. молекулы адсорбата не могут
перемещаться по поверхности адсорбента.
Так как хемосорбция является химическим
процессом, требующим энергии активации
порядка 40 – 120 кДж/моль, повышение
температуры способствует её протеканию.
Примером химической адсорбции является
адсорбция кислорода на вольфраме или
серебре при высоких температурах.
Избирательная
адсорбция-
процесс, основанный на неодинаковой
способности отдельных компонентов
газовой или жидкой смеси поглощаться
из данного растворителя данным
адсорбентом. В аналитической практике
и в технике вариации в выборе адсорбентов
и десорбирующих растворителей
(десорбентов), а также вариации в режиме
процесса позволяют осуществлять самые
различные задачи А. с. Она является
основой метода разделения многокомпонентных
смесей, называемого хроматографией.
Величина
адсорбции зависит от
природы адсорбата, поверхности
адсорбента,
концентрации (давлениях температуры и
др. факторов. Зависимость величины
адсорбции от концентрации адсорбированного
вещества при T=сonst называется изотермой адсорбции.
Адсорбция из предельно разбавленных
растворов подчиняется линейному закону:
,
где K —константа
равновесия, [S − P] и [S*] —
доли поверхности адсорбента, занятые
и незанятые адсорбатом, а [P] —
концентрация адсорбтива.Химическая
адсорбция (хемосорбция)
осуществляется путем химического
взаимодействия молекул адсорбента и
адсорбата. Хемосорбция обычно необратима;
химическая адсорбция, в отличие от
физической, является локализованной,
т.е. молекулы адсорбата не могут
перемещаться по поверхности адсорбента.
Так как хемосорбция является химическим
процессом, требующим энергии активации
порядка 40 – 120 кДж/моль, повышение
температуры способствует её протеканию.
Примером химической адсорбции является
адсорбция кислорода на вольфраме или
серебре при высоких температурах.
Избирательная
адсорбция-
процесс, основанный на неодинаковой
способности отдельных компонентов
газовой или жидкой смеси поглощаться
из данного растворителя данным
адсорбентом. В аналитической практике
и в технике вариации в выборе адсорбентов
и десорбирующих растворителей
(десорбентов), а также вариации в режиме
процесса позволяют осуществлять самые
различные задачи А. с. Она является
основой метода разделения многокомпонентных
смесей, называемого хроматографией.
Величина
адсорбции зависит от
природы адсорбата, поверхности
адсорбента,
концентрации (давлениях температуры и
др. факторов. Зависимость величины
адсорбции от концентрации адсорбированного
вещества при T=сonst называется изотермой адсорбции.
Адсорбция из предельно разбавленных
растворов подчиняется линейному закону:
![]() или
или ![]() (1.12)
(1.12)
где k и k/ - константы.
Для более широкого спектра концентраций изотерма мономолекулярной адсорбции описывается уравнением Ленгмюра;
![]() или
или ![]()
где Г∞ - емкость монослоя или предельная величина адсорбции, представляющая собой число молей ПАВ, приходящихся на 1 единицу поверхности в насыщенном адсорбционном слое;
b, b/- константы адсорбционного взаимодействия, характеризующие энергию адсорбции.
28Дисперсные системы - гетерогенные системы из двух или большего числа фаз с сильно развитой поверхностью раздела между ними. Обычно одна из фаз образует непрерывную дисперсионную среду, в объеме которой распределена дисперсная фаза (или несколько дисперсных фаз) в виде мелких кристаллов, твердых аморфных частиц, капель или пузырьков. По степени раздробленности (дисперсности) системы делятся на следующие классы:
1)грубодисперсные, размер частиц в которых более 10-5 м; 2)тонкодисперсные (микрогетерогенные) с размером частиц от 10-5 до 10-7 м; 3)коллоидно-дисперсные (ультрамикро-гетерогенные)с частицами размером от 10-7до 10-9м.
Если фиксировать внимание на двух основных компонентах дисперсных систем, то одному из них следует приписать роль дисперсионной среды, а другому - роль дисперсной фазы. В этом случае все дисперсные системы можно классифицировать по агрегатным состояниям фаз. По агрегатным состояниям фаз- два класса: свободнодисперсные системы и сплошные (или связнодисперсные) системы .В свободнодисперсных системах дисперсная фаза не образует сплошных жестких структур (сеток, ферм или каркасов). Эти системы называют золями. В сплошных (связнодисперсных) системах частицы дисперсной фазы образуют жесткие пространственные структуры (сетки, каркасы, фермы). Такие системы оказывают сопротивление деформации сдвига. Классификация дисперсных систем по силе межмолекулярного взаимодействия: 1)лиофобные- слабое взаимодействие между дисперсной фазой и дисперсной средой 2)лиофильные- сильное взаимодействие Коллоидными системами называют двух или многофазные системы, в которых одна фаза находится в виде отдельных мелких частиц, распределенных в другой фазе. Такие ультрамикрогетерогенные системы с определенной (коллоидной) дисперсностью проявляют способность к интенсивному броуновскому движению и обладают высокой кинетической устойчивостью. Имея высокоразвитую поверхность раздела фаз и, следовательно, громадный избыток свободной поверхностной энергии, эти системы являются принципиально термодинамически неустойчивыми, что выражается в агрегации частиц, т.е. в отсутствии агрегативной устойчивости. Это системы с очень малой межфазовой энергией, они термодинамически устойчивы и образуются самопроизвольно. Мицеллы (уменьшительное от лат.mica — частица, крупинка) — частицы вколлоидных системах, состоят из нерастворимого в данной среде ядра очень малого размера, окруженного стабилизирующей оболочкой адсорбированных ионов и молекулрастворителя. Например, мицелла сульфида мышьяка имеет строение: {(As2S3)m•nHS−•(n-x)H+}x-•хН+ Средний размер мицелл от 10−5 до 10−7см. К мицеллам относят также частицы в растворах поверхностно-активных веществ (ПАВ), называемых лиофильными коллоидами. Например, мицеллы додецилсульфатав воде. В лиофильных золях мицелла представляет собой ассоциат молекул (агрегаты, состоящие из десятка и сотен амфильных молекул). В каждой молекуле длинный гидрофобный радикал связан с полярной (гидрофильной) группой. При образовании мицеллы несколько десятков или сотен молекул объединяются так, что гидрофобные радикалы образуют ядро (внутреннюю область), а гидрофильные группы — поверхностный слой мицеллы. Минимальную концентрацию поверхностно-активных веществ в растворе, при которой в системе образуются устойчивые мицеллы, находящиеся в равновесии с неассоциированными молекулами поверхностно-активного вещества, называют критической концентрацией мицеллоообразования. Если дисперсионной средой является органическая жидкость, ориентация молекул в мицелле может быть обратной: ядро содержит полярные группы, а гидрофобные радикалы обращены во внешнюю фазу (обратная мицелла)[1]. Мицеллы могут существовать в состояниях с различными равновесными структурами и в различных внешних формах, устойчивых при различных концентрациях ПАВ в мицеллярном растворе. Такая способность мицелл называется полиморфизмом мицелл. В лиофобных гидрозолях, стабилизованных электролитами, ядро мицеллы окружено двумя слоями противоположно заряженных ионов, то естьдвойным электрическим слоем. Диффузный слой ионов препятствует сближению и агрегированию (сцеплению) частиц.
![]()
29.Оптические
свойства: рассеивание света.
Электрокинетические свойства: электроосмос
и электрофорез. Строение двойного
электрического слоя.Выделяют 5 типов
взаимодействия электромагнитного
излучения с изучаемой системой:
пропускание, отражение, рассеивание,
преломление и поглощение. Для истинных
расстоворо характерно пропускание, они
являются оптически прозрачными.
Грубодисперсные системы называют
оптически мутными, так как размеры
частиц этих систем много больше длины
волны электромагнитного излучения. Для
золей характерно рассеивание света.
Рассеивание происходит в системах,
размер частиц которых не превышает
0,1-0,2 длины волны падающего света. В золях
возникает дифракционное рассеивание
в результате огибания частиц световой
волной. Визуально наблюдается опалесценция
– различие окраски в проходящем и
рассеивающемся свете (наблюдаемое
явление носит название конуса Тиндаля
в честь физика, открывшего рассеивание
света) По наличию конуса Тиндаля можно
отличить коллоидные растворы от истинных.
Закон светорассеивания Рэлея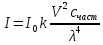 , гдеI-интенсивность
рассеянного света,
, гдеI-интенсивность
рассеянного света,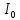 -
интенсивность падающего света,k-
константа Рэлея (зависит от показателей
преломления дисперсионной среды и
фазы),V-объем частицы
дисперсионной фазы, λ – длина волны
падающего света,
-
интенсивность падающего света,k-
константа Рэлея (зависит от показателей
преломления дисперсионной среды и
фазы),V-объем частицы
дисперсионной фазы, λ – длина волны
падающего света,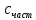 -
частичная концентрация. Выводы
Рэлея: 1) интенсивность рассеянного
света прямо пропорциональна концентрации
частиц. 2) интенсивность рассеянного
света прямо пропорциональна квадрату
объема частицы. 3) в большей степени при
прохождении через золи рассеивается
коротковолновое излучение. Электрофорез
– движение частиц в электрическом поле,
т.е. перемещение частиц дисперсной фазы
относительно неподвижной дисперсионной
среды под действие внешнего электрического
поля. Электрофорез применяется для
разделения аминокислот и белков,
нуклеиновых кислот, антибиотиков,
ферментов, антител, форменных элементов
крови и т.д. В клинических исследованиях
электрофорез используют для диагностики
заболеваний, сопровождающихся изменением
состава белков. Электроосмос – движение
растворителя в электрическом поле, т.е.
перемещение дисперсионной среды
относительно неподвижной дисперсной
фазы в электрическом поле. Электроосмос
и электрофорез обусловлены наличием
двойного электрического поля на
поверхности частицы золя. Двойной
электрический слой состоит возникает
на границе раздела фаз: твердое вещество
– раствор электролита. Он состоит из
достаточно прочно связанных с поверхностью
дисперсной фазы потенциалопределяющих
ионов и противоионов, находящихся в
дисперсной среде. Потенциалобразующие
ионы вместе со связанными противоионами
образуют адсорбционный слой. Возникает
разность потенциалов. Строение двойного
электрического слоя: 1 – потенциалопределяющие
ионы 2 – адсорбционный слой противоионов
3 – диффузный слой противоионов АВ-
плоскость скольжения
-
частичная концентрация. Выводы
Рэлея: 1) интенсивность рассеянного
света прямо пропорциональна концентрации
частиц. 2) интенсивность рассеянного
света прямо пропорциональна квадрату
объема частицы. 3) в большей степени при
прохождении через золи рассеивается
коротковолновое излучение. Электрофорез
– движение частиц в электрическом поле,
т.е. перемещение частиц дисперсной фазы
относительно неподвижной дисперсионной
среды под действие внешнего электрического
поля. Электрофорез применяется для
разделения аминокислот и белков,
нуклеиновых кислот, антибиотиков,
ферментов, антител, форменных элементов
крови и т.д. В клинических исследованиях
электрофорез используют для диагностики
заболеваний, сопровождающихся изменением
состава белков. Электроосмос – движение
растворителя в электрическом поле, т.е.
перемещение дисперсионной среды
относительно неподвижной дисперсной
фазы в электрическом поле. Электроосмос
и электрофорез обусловлены наличием
двойного электрического поля на
поверхности частицы золя. Двойной
электрический слой состоит возникает
на границе раздела фаз: твердое вещество
– раствор электролита. Он состоит из
достаточно прочно связанных с поверхностью
дисперсной фазы потенциалопределяющих
ионов и противоионов, находящихся в
дисперсной среде. Потенциалобразующие
ионы вместе со связанными противоионами
образуют адсорбционный слой. Возникает
разность потенциалов. Строение двойного
электрического слоя: 1 – потенциалопределяющие
ионы 2 – адсорбционный слой противоионов
3 – диффузный слой противоионов АВ-
плоскость скольжения
![]()
30. Электрокинетический потенциал и его зависимость от различных факторов. Устойчивость дисперсных систем. Устойчивость коллоидных систем. Коагуляция. Порог коагуляции и его определение. Коллоидная защита и пептизация. Коагуляция в биологических системах. Скачок потенциалов на поверхности скольжения называется дзета-потенциалом, или электрокинетическим потенциалом. Его значение определяется толщиной диффузного слоя и числом противоионов в диффузном слое(чем меньше толщина слоя, тем меньше потенциал).Его рассчитывают по уравнению: ξ = ƞ*h*L/ Ɛ*Ɛ0*U*Ʈ где ƞ – вязкость дисперсионной среды h – смещение границ, м L – расстояния между концами агаровых сифонов, м Ɛ – диэлектрическая проницаемость воды (80) Ɛ0 – электрическая постоянная, равная 8,85*10-12 Ф/м U – разность потенциалов на электродах, В. Ʈ – время, с. Устойчивость дисперсных систем характеризуется постоянством дисперсности (распределения частиц по размерам) и концентрации дисперсной фазы (числом частиц в единице объема). Различают кинетическую и агрегативную устойчивость. Кинетическая устойчивость обусловлена диффузией и броуновским движением коллоидных частиц, которые препятствуют оседанию частиц под действием силы тяжести. Агрегативная устойчивость – способность системы к сохранению степени дисперсности. Причиной агрегативной устойчивости является наличие у коллоидных частиц электрического заряда, который препятствует слипанию частиц, а также способствует образованию развитых сольватных защитных слоев. Потеря агрегативной устойчивости происходит путем коагуляции – укрупнения коллоидных частиц. Она может быть вызвана: действием тепла, излучений, электролитов или другого коллоидного раствора с частицами противоположного заряда. Различают скрытую и явную коагуляцию. При скрытой коагуляции происходит некоторое уменьшение общего числа коллоидных частиц, но осадок не выпадает, видимых изменений нет. В результате скрытой коагуляции происходит изменение вязкости золя, степени дисперсности. При явной коагуляции каждое соударение частиц приводит к слипанию, образуется осадок. Наименьшая концентрация электролита, которая может вызвать явную коагуляцию золя, называется пороговой и характеризует порог коагуляции (от 10-5 до 0,1 моль в литре золя). Определение порогов коагуляции визуальным методом: Берут растворы NaCl, BaCI2. Методом двойных разбавлений готовят по 5 растворов каждого электролита ( в пробирку отбирают 1 мл раствора электролита и 1 мл дистиллированной воды, затем из образующегося раствора отбирают 1 мл и смешивают в другой пробирке с 1 мл дистиллированной воды и т.д.) В пробирку отбирают 1 мл латекса и 1 мл раствора электролита, встряхивают и через 5-10 мин отмечают эффект коагуляции. Сравнив найденные значения порогов коагуляции электролитов, определяют знак иона-коагулятора и, соответственно, знак заряда коллоидных частиц исследуемого золя. Более точно определение порогов коагуляции можно осуществить с помощью фотоэлектроколориметра. В этом случае изменение дисперсности в результате коагуляции обнаруживают по изменению оптических свойств системы, в частности по изменению интенсивности светорассеивания. Основные причины коагуляции под действием электролитов вызваны уменьшение дзета-потенциала вследствие: 1) сжатия диффузного слоя 2) адсорбции на коллоидной частице ионов добавленного электролита, имеющих заряд, противоположный заряду гранул. Пептизацией называют процесс перехода свежеполученного при коагуляции осадка в золь под действием пептизаторов. Пептизация – процесс, обратный коагуляции. Пептизаторами могут быть электролиты и неэлектролиты. Различают адсорбционную и химическую пептизацию. Примером адсорбционной пептизации может служить переход в золь свежеполученного и промытого водой осадка гидроксида железа(3) при добавлении к нему небольших количеств раствора хлорида железа(3). Пример химической пептизации: пептизация осадка гидроксида железа (3) соляной кислотой. Происходит химическая реакция соляной кислоты с частью осадка, ионы пептизаторы адсорбируются на частицах осадка. Процессы коагуляции имеют большое значение в жизнедеятельности организма. Для сохранения постоянства физико-химических условий в организме необходимо соблюдать постоянство не только концентрации электролитов, но и их качественного состава. Например, изотонический раствор MgSO4 будет обладать более сильным коагулирующим действием, чем раствор NaCl. Часто необходимо стабилизировать коллоидный раствор. Такой способностью обладают некоторые ПАВ, например, желатин и другие белки, полисахариды, пектиновые вещества, обладающие так называемым защитным действием. Коллоидная защита играет важную роль в ряде физиологических процессов. Белки крови являются защитой для жира, холестерина. В крове и моче содержатся в коллоидном состоянии труднорастворимые фосфат, карбонат, оксалат кальция, стабилизированные защитными веществами белкового характера. При некоторых заболеваниях содержание защитных веществ уменьшается, что приводит к выпадению указанных солей в осадок (образование камней в почках, печени, отложение солей в суставах и др.) Измерением защитного числа («золотого числа») спинномозговой жидкости пользуются для диагностики некоторых заболеваний, например, менингита. Лекарственные препараты бактерицидного действия – протаргол и колларгол – являются золями металлического серебра, защищенными белками.