

Образование связи
Простая ковалентная связь образуется из двух неспаренных валентных электронов, по одному от каждого атома:
![]()
В результате обобществления электроны образуют заполненный энергетический уровень. Связь образуется, если их суммарная энергия на этом уровне будет меньше, чем в первоначальном состоянии (а разница в энергии будет не чем иным, как энергией связи).
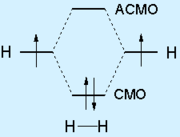
![]()
Заполнение электронами атомных (по краям) и молекулярных (в центре) орбиталей в молекуле H2. Вертикальная ось соответствует энергетическому уровню, электроны обозначены стрелками, отражающими их спины.
Согласно теории молекулярных орбиталей, перекрывание двух атомных орбиталей приводит в простейшем случае к образованию двух молекулярных орбиталей (МО): связывающей МО и антисвязывающей (разрыхляющей) МО. Обобществленные электроны располагаются на более низкой по энергии связывающей МО.
Виды ковалентной связи
Существуют три вида ковалентной химической связи, отличающихся механизмом образования:
1. Простая ковалентная связь. Для ее образования каждый из атомов предоставляет по одному неспаренному электрону. При образовании простой ковалентной связи формальные заряды атомов остаются неизменными.
Если атомы, образующие простую ковалентную связь, одинаковы, то истинные заряды атомов в молекуле также одинаковы, поскольку атомы, образующие связь, в равной степени владеют обобществленной электронной парой. такая связь называется неполярной ковалентной связью. Такую связь имеют простые вещества, например О2; N2; C12.
Если атомы различны, то степень владения обобществленной парой электронов определяется различием в электроотрицательностях атомов. Атом с большей электроотрицательностью сильнее притягивает к себе пару электронов связи, и его истинный заряд становится отрицательным. Атом с меньшей электроотрицательностью приобретает, соответственно, такой же по величине положительный заряд. Такая ковалентная связь называется полярной.
2. Донорно-акцепторный механизм. Для образования этого вида ковалентной связи оба электрона предоставляет один из атомов — донор. Второй из атомов, участвующий в образовании связи, называется акцептором. В образовавшейся молекуле формальный заряд донора увеличивается на единицу, а формальный заряд акцептора уменьшается на единицу.
3. Семиполярная связь. Этот вид ковалентной связи образуется между атомом, обладающим неподеленной парой электронов (азот, фосфор, сера, галогены и т. п.) и атомом с двумя неспаренными электронами (кислород, сера). Образование семиполярной связи протекает в два этапа:
1. Окисление (перенос одного электрона) атома с НЭП атомом с двумя неспаренными электронами. В результате атом с НЭП превращается в катион-радикал (положительно заряженная частица с неспаренным электроном), а атом с двумя неспаренными электронами — в анион-радикал (отрицательно заряженная частица с неспаренным электроном).
2. Обобществление неспаренных электронов (как в случае простой ковалентной связи).
При образовании семиполярной связи атом с НЭП увеличивает свой формальный заряд на единицу, а атом с двумя неспаренными электронами понижает свой формальный заряд на единицу.
Одно из существенных свойств ковалентной связи - ее насыщаемость. При ограниченном числе внешних электронов в областях между ядрами образуется ограниченное число электронных пар вблизи каждого атома (и, следовательно, число химических связей). Именно это число тесно связано с понятием валентности атома в молекуле (валентностью называют общее число ковалентных связей, образуемых атомом). Другое важное свойство ковалентной связи - ее направленность в пространстве. Это проявляется в примерно одинаковом геометрическом строении близких по составу химических частиц. Особенностью ковалентной связи является также ее поляризуемость
В большинстве присланных работ дан неполный ответ на задание 3: "Химическая связь между атомами в молекулах образуется с помощью электронных пар, которые принадлежат обоим атомам. Поэтому большинство молекул содержат четное количество электронов. Имеются, однако, молекулы с нечетным числом электронов. Приведите примеры таких молекул. Как можно объяснить образование химической связи в молекулах с нечетным числом электронов?". В частности, недостаточно представлено примеров молекул с нечетным числом электронов. В основном рассматривались оксиды азота и хлора. Существует таже значительное количество молекул, образованных d- и f-элементами с нечетным числом электронов: оксид меди(II), оксиды марганца (II),(IV),(VI), хлорид железа(III) и др. Многие забыли о существовании органических молекул с нечетным числом электронов. Образование химической связи в ковалентных молекулах объясняется с использованием метода молекулярных орбиталей (что представлено в некоторых работах участников олимпиады). А о том, что неспаренные что d-электроны могут не принимать участие в образовании химической связи и полученная молекула окажется с нечетным числом электронов, написано лишь в одной работе!
34.
Скорость химической реакции — изменение количества вещества одного из реагирующих веществ за единицу времени в единице реакционного пространства. Является ключевым понятием химической кинетики. Скорость химической реакции — величина всегда положительная, поэтому, если она определяется по исходному веществу (концентрация которого убывает в процессе реакции), то полученное значение домножается на −1.
Например для реакции:
![]()
выражение для скорости будет выглядеть так:
![]() .
.
В 1865 году Н. Н. Бекетовым и в 1867 году Гульдбергом и Вааге был сформулирован закон действующих масс:
Скорость химической реакции в каждый момент времени пропорциональна концентрациям реагентов, возведенным в некоторые степени.
Для элементарных реакций показатель степени при значении концентрации каждого вещества часто равен его стехиометрическому коэффициенту, для сложных реакций это правило не соблюдается. Кроме концентрации на скорость химической реакции оказывают влияние следующие факторы:
-
природа реагирующих веществ,
-
наличие катализатора,
-
температура (правило Вант-Гоффа),
-
давление,
-
площадь поверхности реагирующих веществ.
Если мы рассмотрим самую простую химическую реакцию A + B → C , то мы заметим, что мгновенная скорость химической реакции величина непостоянная.
Прямое измерение скорости гомогенной реакции достигается с помощью проточного перемешиваемого реактора. В сосуд, снабженный мощной мешалкой, с постоянной скоростью вводят исходные вещества и выводят реагирующую смесь так, чтобы её количество в реакционном сосуде было постоянно. При установившемся стационарном состоянии анализ отбираемой смеси показывает состав реагирующей смеси. Зная, кроме того, скорость отбора этой смеси, определяют количество вещества, образовавшегося в результате реакции за единицу времени, а отсюда — С. х. р. Для гетерогенно-каталитических процессов с неподвижным катализатором эквивалентом описанного метода является проточно-циркуляционный метод: однородность состава реагирующей смеси в зоне реакции достигается с помощью создаваемой насосом интенсивной циркуляции реагирующей смеси. Проточные перемешиваемые реакторы и проточно-циркуляционные системы принадлежат к классу безградиентных реакторов, называемых так потому, что в них практически отсутствуют градиенты (перепады) концентраций, а также температуры в зоне реакции.
Особые трудности возникают при изучении очень быстрых реакций в растворах. Если реакция успевает пройти в значительной степени за время, которое требуется для смешения растворов исходных веществ, то обычные методы непригодны. Задача измерения скоростей таких реакций решается с помощью релаксационных методов, разработанных М. Эйгеном. Система, в которой может происходить обратимая реакция, вначале находится в состоянии равновесия химического. Затем весьма быстро изменяют параметр, влияющий на значение константы равновесия: температуру, давление или электрическое поле. Система переходит к новому состоянию равновесия в течение некоторого времени; этот процесс называется релаксацией. Следя за изменением состава каким-либо безынерционным методом (например, по электропроводности), определяют С. х. р. Удаётся наблюдать время релаксации до 10-6 сек: таким путём была измерена, например, скорость реакции Н++ OH- = H2O в воде.
Влияние концентраций реагирующих веществ. Чтобы осуществлялось химическое взаимодействие веществ А и В, их молекулы (частицы) должны столкнуться. Чем больше столкновений, тем быстрее протекает реакция. Число же столкновений тем больше, чем выше концентрация реагирующих веществ. Отсюда на основе обширного экспериментального материала сформулирован основной закон химической кинетики, устанавливающий зависимость скорости реакции от концентрации реагирующих веществ:
Cкорость химической реакции пропорциональна произведению концентраций реагирующих веществ.
Для реакции ( I ) этот закон выразится уравнением
v = kcA cB , (1)
где сА и сВ - концентрации веществ А и В, моль/л; k - коэффициент пропорциональности, называемый константой скорости реакции. Основной закон химической кинетики часто называют законом действующих масс.
Из уравнения (1) нетрудно установить физический смысл константы скорости k : она численно равна скорости реакции, когда концентрации каждого из реагирующих веществ составляют 1 моль/л или когда их произведение равно единице.
Константа скорости реакции k зависит от природы реагирующих веществ и от температуры, но не зависит от их концентраций.
Уравнение (1), связывающее скорость реакции с концентрацией реагирующих веществ, называется кинетическим уравнением реакции. Если опытным путем определено кинетическое уравнение реакции, то с его помощью можно вычислять скорости при других концентрациях тех же реагирующих веществ.
Влияние температуры .
Зависимость скорости реакции от температуры определяется правилом Вант-Гоффа:
При повышении температуры на каждые 10о скорость большинства реакций увеличивается в 2-4 раза.
Математически эта зависимость выражается соотношением
![]()
vt 2 = vt 1 γ ,
где vt 1 , vt 2 - скорости реакции соответственно при начальной ( t 1 ) и конечной ( t 2 ) температурах, а γ - температурный коэффициент скорости реакции, который показывает, во сколько раз увеличивается скорость реакции с повышением температуры реагирующих веществ на 10°.
Правило Вант-Гоффа является приближенным и применимо лишь для ориентировочной оценки влияния температуры на скорость реакции. Температура влияет на скорость химической реакции, увеличивая константу скорости.
36.
Окисли́тельно-восстанови́тельные реа́кции — это химические реакции, протекающие с изменением степеней окисления атомов, входящих в состав реагирующих веществ, реализующихся путём перераспределения электронов между атомом-окислителем и атомом-восстановителем.
37.
Длина связи в молекуле - это расстояние между ядрами соседних атомов, образующих связь (обычно порядка 0.1-0.3 нм). Приближенно длина связи равна сумме радиусов соседних атомов. Как и радиусы атомов межъядерные расстояния закономерно изменяются в рядах, подгруппах Периодической системы. Расстояние между одинаковыми атомами в различных соединениях (при одинаковой кратности связи) близки. Чем выше кратность связи, тем меньше ее длина и больше энергия связи, а при одинаковой кратности - чем меньше длина, тем больше энергия связи.
|
Например, кратность связи "СО" в ионе СО32- составляет примерно 11/3 (т.е. в редких случаях может быть и дробной). Длина связи "С-О" в ионе СО32- равна 0.129 (нм) и является промежуточной между длиной одинарной (0.143) и двойной (0.122 нм) связей. |
Длина кратной связи уменьшается с увеличением ее кратности. Например, в атоме азота существует тройная связь (6 электронов являются общими: по 3 общих электрона с каждой стороны). Энергия химических связей - это энергия (кДж/моль), которая выделяется при образовании (1 моль) связи или поглощается при разрыве (1 моль) связи. Чем больше энергия связи, тем прочнее связь. Любая химическая реакция заключается в разрыве одних химических связей и образовании других. При этом из молекулы образуются атомы, радикалы, ионы или возбужденные молекулы. Чем больше перекрывание орбиталей атомов, тем больше энергия связи и тем прочнее химическая связь. Энергия ковалентных связей обычно составляет 100-500 кДж/моль, энергия водородных связей - 20-40 кДж/моль. Для справки, энергия разрыва молекулы: "Н" равна EH-H = 432 кДж/моль; "О" равна Eo=o = 494 кДж/моль. В нижеприведенных реакциях: H2 <==> H+ + H+ (ECB = 432 кДж/моль) H2O <==> H+ + OH+ (ECB = 461 кДж/моль) C2H6 <==> .CH3 + .CH3 (ECB = 356 кДж/моль) энергия выделяется. Энергия связи зависит от продуктов, которые получаются в результате ее разрыва. При разрыве одинаковых связей энергия изменяется - постепенно увеличивается (например, при последовательном отрыве атомов "H" от "CH4"). Связь между двумя одинаковыми атомами может обладать различными энергиями (например, связь между атомами азота имеет энергию около 160 кДж/моль в N2H4 и более 900 - в N2). Атомы не соединялись бы между собой с образованием молекул, если бы это не вело к "выигрышу" (то есть высвобождению) энергии. Зависимость энергии и длины связи от кратности: - при одинарной связи (ее длина равна 154 нм, энергия -356 кДж/моль) - при двойной связи (ее длина равна 134 нм, энергия - 598 кДж/моль) - при тройной связи (ее длина равна 120 нм, энергия - 813 кДж/моль) Ковалентный радиус - это половина расстояния между ядрами атомов данного элемента, образующими ковалентную связь. Например, для серы и селена - половина длины связи в молекуле X8, а для углерода он принимается равным половине длины связи в решётке алмаза, и т.п. Ковалентный радиус характеризует распределение электронной плотности вблизи ядра. Ковалентный радиус близок численно и по смыслу к другим характеристикам распределения электронной плотности. Полярность ковалентной связи - определяется смещением общей электронной пары к одному из атомов. Количественно она выражается через дипольный момент m (m = |q|*l). Чем больше "m", тем более полярна связь. Абсолютное значение дипольного момента ("m") определяют экспериментально. Единицей измерения дипольных моментов молекул служит дебай(1 Дебай = 3,33*10-30 Кл*м). У большинства молекул дипольные моменты меньше этой величины. Это обстоятельство означает, что реально существующие на атомах заряды обычно меньше одного элементарного заряда (т.е. нецелочисленны).
|
Дипольные моменты сложных молекул получаются путем сложения дипольных моментов отдельных связей по правилам сложения векторов. В случае симметричных фигур дипольный момент молекулы с полярными связями оказывается равным нулю, поэтому молекула СO2 линейна: "O=C=O". Линейны и более сложные симметричные молекулы: SiCl4, SF6, CO3 2- и др. По величине дипольного момента можно сделать заключение как о характере химической связи (ионная, ковалентная полярная или неполярная), так и о геометрической форме молекулы. |
Насыщаемость связи - это
способность атома соединяться с другим
атомом определенным числом связей,
которое определяется числом валентных
электронов (именно это число тесно
связано с традиционным понятием
валентности атома в молекуле). Насыщаемость
проявляется в том, что одна орбиталь
атома с неспаренным электроном может
принимать участие в образовании только
одной ковалентной химической связи.
Направленность связи - определяется
величиной угла между направлениями
связей в пространстве (направленностью
в пространстве перекрывающихся атомных
орбиталей). Это свойство проявляется в
примерно одинаковом строении родственных
по составу молекулярных фрагментов
(например, фрагмент СН2 в различных
углеводородах имеет примерно одно и то
же строение). Перекрывание электронных
облаков может осуществляться двумя
путями:
а) Если электронные облака
перекрываются по линии, соединяющей
центры этих облаков, то образующаяся
при этом связь называется (сигма)-связью.
Чем больше перекрывание облаков, тем
прочнее связь:
 (сигма)s-
связь
(сигма)s-
связь
![]() (сигма)p-
связь
б) Если электронные облака
перекрываются по обе стороны от линии,
соединяющей центры облаков, то такая
связь называется (пи) связью.
(сигма)p-
связь
б) Если электронные облака
перекрываются по обе стороны от линии,
соединяющей центры облаков, то такая
связь называется (пи) связью.
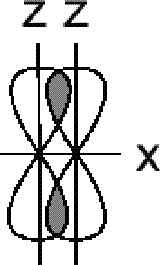 (пи)pz-
связь
(пи)pz-
связь
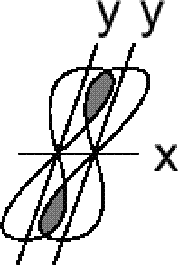 (пи)py-
связь
Ионная (или электровалентная)
связь - это химическая связь,
обусловленная образованием электронных
пар за счет перехода валентных электронов
от одного атома (металла) к другому атому
(неметаллу) в результате электростатического
притяжения. Атомы, отдающие свои
электроны, превращаются в положительно
заряженные ионы (катионы) и проявляют
положительную валентность. Атомы,
принимающие электроны, превращаются в
отрицательно заряженные анионы. Такая
связь возникает между ионами и является
частным случаем полярной ковалентной
связи: электронная пара, образующая
связь, полностью смещена к одному из
атомов.
Существование молекулы как
единого целого в случае ионной связи
обеспечивается электростатическим
взаимодействием образовавшихся ионов.
Резкой границы между соединениями с
полярной ковалентной и ионной связями
не существует. Не существует и идеально
ионных соединений. Даже при химическом
взаимодействии наиболее электроположительных
и электроотрицательных элементов
образуются сильно полярные соединения
с наибольшей долей ионности.
Валентность
элементов в ионных соединениях равна
заряду иона. Заряд иона указывается
вверху справа от химического знака,
например: Al3+, Ca2+, S2- и
т.д.
Например, при взаимодействия
металла "Na" и неметалла "Cl"
электроны с внешней оболочки атома Na
переходит к атому "Cl":
"Na" -
"e" = Na+
"Cl" + "e" =
Cl-
Между ионами натрия и хлора
возникают электростатические силы
притяжения,образуется молекула "NaCl",в
которой связь между ионами ионная.
Ионные соединения образуются атомами
элементов, имеющими большую разность
в электроотрицательности, т.е. это, как
правило, связь между атомами элементов
главных подгрупп I, II и VI, VII групп
периодической системы. К ионным
соединениям близки только галогениды
щелочных металлов, хотя и для них
эффективные заряды не достигают единицы.
Так как образование связи между
атомами в молекуле происходит за счет
возникновения общих электронных пар,
то при этом атомы, обладающие большей
способностью притягивать электроны на
свою внешнюю оболочку (более
электроотрицательные), будут приобретать
избыточный отрицательный заряд, а менее
электроотрицательные - положительный
заряд, например:
Na+---Cl-
Разность в значениях электроотрицательности
связанных атомов в этом случае имеет
значение больше двух. Случаю предельной
поляризации отвечает идеальная ионная
связь.
Молекулы с ионной связью
существуют только в парообразном
состоянии. При обычной температуре все
ионные соединения - это ионные кристаллы,
в которых нет отдельных молекул (например,
ионы "NaCl" это как бы одна молекула,
состоящая из чередующихся положительных
и отрицательных ионов). Все вещества с
ионной связью хрупки, плохо проводят
тепло, не проводят электрический ток
(например, если соль насыпать на сковородку
и нагревать, то сковорода нагревается
быстро, а соль - медленно).
(пи)py-
связь
Ионная (или электровалентная)
связь - это химическая связь,
обусловленная образованием электронных
пар за счет перехода валентных электронов
от одного атома (металла) к другому атому
(неметаллу) в результате электростатического
притяжения. Атомы, отдающие свои
электроны, превращаются в положительно
заряженные ионы (катионы) и проявляют
положительную валентность. Атомы,
принимающие электроны, превращаются в
отрицательно заряженные анионы. Такая
связь возникает между ионами и является
частным случаем полярной ковалентной
связи: электронная пара, образующая
связь, полностью смещена к одному из
атомов.
Существование молекулы как
единого целого в случае ионной связи
обеспечивается электростатическим
взаимодействием образовавшихся ионов.
Резкой границы между соединениями с
полярной ковалентной и ионной связями
не существует. Не существует и идеально
ионных соединений. Даже при химическом
взаимодействии наиболее электроположительных
и электроотрицательных элементов
образуются сильно полярные соединения
с наибольшей долей ионности.
Валентность
элементов в ионных соединениях равна
заряду иона. Заряд иона указывается
вверху справа от химического знака,
например: Al3+, Ca2+, S2- и
т.д.
Например, при взаимодействия
металла "Na" и неметалла "Cl"
электроны с внешней оболочки атома Na
переходит к атому "Cl":
"Na" -
"e" = Na+
"Cl" + "e" =
Cl-
Между ионами натрия и хлора
возникают электростатические силы
притяжения,образуется молекула "NaCl",в
которой связь между ионами ионная.
Ионные соединения образуются атомами
элементов, имеющими большую разность
в электроотрицательности, т.е. это, как
правило, связь между атомами элементов
главных подгрупп I, II и VI, VII групп
периодической системы. К ионным
соединениям близки только галогениды
щелочных металлов, хотя и для них
эффективные заряды не достигают единицы.
Так как образование связи между
атомами в молекуле происходит за счет
возникновения общих электронных пар,
то при этом атомы, обладающие большей
способностью притягивать электроны на
свою внешнюю оболочку (более
электроотрицательные), будут приобретать
избыточный отрицательный заряд, а менее
электроотрицательные - положительный
заряд, например:
Na+---Cl-
Разность в значениях электроотрицательности
связанных атомов в этом случае имеет
значение больше двух. Случаю предельной
поляризации отвечает идеальная ионная
связь.
Молекулы с ионной связью
существуют только в парообразном
состоянии. При обычной температуре все
ионные соединения - это ионные кристаллы,
в которых нет отдельных молекул (например,
ионы "NaCl" это как бы одна молекула,
состоящая из чередующихся положительных
и отрицательных ионов). Все вещества с
ионной связью хрупки, плохо проводят
тепло, не проводят электрический ток
(например, если соль насыпать на сковородку
и нагревать, то сковорода нагревается
быстро, а соль - медленно).
|
Ионная связь является ненаправленной и ненасыщаемой. Ненаправленность ионной связи заключается в том, что каждый ион создает вокруг себя электрическое поле, силовые линии которого направлены во все стороны от него. Поэтому любой положительный ион притягивает к себе множество отрицательных ионов, образуя кристалл. Никогда нельзя определить направление ионной связи: она действует сразу во всех направлениях. Например, в кристалле "NaCl" каждый ион натрия окружен восемью ионами хлора, и каждый ион хлора окружен восемью ионами натрия. Практически даже при образовании связи между такими сильно противоположными по свойствам элементами, как натрия и хлора, нет полного отрыва электронного облака: оно только в большей степени смещено к атому хлора. Для "NaCl" положительно заряженный ион натрия "Na+" равномерно притягивается всеми отрицательными ионами хлора "Cl-", окружающими его в пространстве, поэтому ионная связь менее прочная, чем ковалентная. |
Металлическая связь - это связь между атомами (положительными ионами) в кристаллической решетке металла, осуществляемая за счет свободно перемещающихся обобществленных электронов. Металл легко отдает свои внешние электроны, например: "Fe" - "2e" <==> Fe+2 Ее свойства напоминают собой как ионную, так и ковалентную полярную связь. Электроны металлов достаточно слабо связаны со своими ядрами и небольшая часть атомов (3-5%) теряют свои валентные электроны (обычно с "s"-подуровня). Облака свободных электронов перекрываются, образуя одно общее электронное облако ("электронный газ", определяющий электропроводность металла), занимающее весь объем кристалла.
|
Электронный газ - совокупность свободных электронов в кристаллической решетке металлов, принадлежащих всем атомам металла, и связывающая их между собой, образуя как бы одну огромную молекулу. |
В любой момент времени существует
динамическое равновесие между атомами,
катионами металла и электронами
(электроны, отрываясь от какого-нибудь
атома, превращают его в катион, но в то
же время другой катион, притягивая
электроны, становится опять нейтральным).
Это облако, в целом заряженное
отрицательно, удерживает в кристалле
катионы металлов, поэтому металл всегда
содержит:
- ряд положительных ионов
(расположенных в определенных положениях
кристаллической решетки);
- большое
количество электронов (свободно
перемещающихся по всему кристаллу).
В
узлах кристаллических решеток находятся
атомы металлов в особом состоянии
(например, в решетке натрия один внешний
электрон легко отрывается от каждого
атома натрия, но эти электроны не
объединяются в пары, а образуют электронный
газ). Электроны в металле осуществляют
связь между всеми атомами металла.
Металлическая связь встречается во
всех металлах, а также в некоторых
карбидах.
Смешанные связи. Если
в состав молекулы входят не 2, а большее
число атомов, то связь между ними может
быть различной. Например, в молекуле
Na2SO4
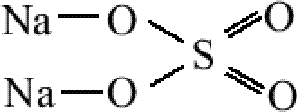 связь
между атомами натрия и кислорода -
ионная, а между кислородом и серой -
ковалентная полярная. Наличие ионной
связи в таких молекулах определяет
физические свойства веществ - Na2SO4
при обычной температуре твердое вещество.
В большинстве случаев при
непосредственном соединении элементов
I и II групп периодической системы с
элементами VI и VII групп получаются ионные
соединения с высокой температуре
плавления и кипения, расплавы которых
проводят электрический ток. Следует
заметить, что в хлористом натрии NaCl
положительно заряженный ион натрия
притягивает к себе отрицательно
заряженный ион хлора не только своей
молекулы, но и других молекул NaCl. Такое
взаимодействие приводит к тому, что
фактически молекулы существуют только
лишь в газе, когда вероятность столкновения
между молекулами мала. В твердом веществе
не существует отдельных молекул NaCl, так
как силы взаимодействия каждого иона
со всеми ионами, которые его окружают,
одинаковы. Поэтому говорят, что данное
твердое вещество состоит из ионов.
Водородная связь - зто связь
между положительно поляризованным
атомом водорода одной молекулы и сильно
отрицательно поляризованным атомом
другой молекулы (например, "N", "O",
"F", реже "Cl", "S"). Электрон
атома водорода в этом случае смещается
в сторону атома другой молекулы,
вследствие чего на атоме водорода
возникает эффективный положительный
заряд, а отсутствие внутренних электронных
слоев у атома водорода позволяет другому
атому сближаться до расстояний, близких
к длинам атомных связей. Полупустая
орбиталь атома водорода частично
присоединяет пару электронов атома
другой молекулы. Водородная связь
сильнее обычного притяжения молекул,
поэтому разрушить молекулы вещества с
водородной связью очень трудно, а
оторвать их друг от друга значительно
легче. Водородная связь может возникнуть:
между двумя фрагментами одной молекулы
- внутримолекулярная связь;
между
двумя разными молекулами - межмолекулярная
связь (эта связь является частным
случаем химической связи).
Образование
водородной связи в случае воды может
быть представлено следующим образом:
связь
между атомами натрия и кислорода -
ионная, а между кислородом и серой -
ковалентная полярная. Наличие ионной
связи в таких молекулах определяет
физические свойства веществ - Na2SO4
при обычной температуре твердое вещество.
В большинстве случаев при
непосредственном соединении элементов
I и II групп периодической системы с
элементами VI и VII групп получаются ионные
соединения с высокой температуре
плавления и кипения, расплавы которых
проводят электрический ток. Следует
заметить, что в хлористом натрии NaCl
положительно заряженный ион натрия
притягивает к себе отрицательно
заряженный ион хлора не только своей
молекулы, но и других молекул NaCl. Такое
взаимодействие приводит к тому, что
фактически молекулы существуют только
лишь в газе, когда вероятность столкновения
между молекулами мала. В твердом веществе
не существует отдельных молекул NaCl, так
как силы взаимодействия каждого иона
со всеми ионами, которые его окружают,
одинаковы. Поэтому говорят, что данное
твердое вещество состоит из ионов.
Водородная связь - зто связь
между положительно поляризованным
атомом водорода одной молекулы и сильно
отрицательно поляризованным атомом
другой молекулы (например, "N", "O",
"F", реже "Cl", "S"). Электрон
атома водорода в этом случае смещается
в сторону атома другой молекулы,
вследствие чего на атоме водорода
возникает эффективный положительный
заряд, а отсутствие внутренних электронных
слоев у атома водорода позволяет другому
атому сближаться до расстояний, близких
к длинам атомных связей. Полупустая
орбиталь атома водорода частично
присоединяет пару электронов атома
другой молекулы. Водородная связь
сильнее обычного притяжения молекул,
поэтому разрушить молекулы вещества с
водородной связью очень трудно, а
оторвать их друг от друга значительно
легче. Водородная связь может возникнуть:
между двумя фрагментами одной молекулы
- внутримолекулярная связь;
между
двумя разными молекулами - межмолекулярная
связь (эта связь является частным
случаем химической связи).
Образование
водородной связи в случае воды может
быть представлено следующим образом:
 Здесь
в структурной формуле точками (пунктирный
штрих) показана водородная связь. Из-за
существенной разницы в электроотрицательности
водорода и кислорода(2.10 и 3.50), атом
кислорода в молекуле воды Н2О
сильно смещает к себе общие электронные
пары, в результате получается избыточный
отрицательный заряд на атоме кислорода
и положительный - на атоме водорода.
Когда две молекулы воды Н2О
оказываются рядом, атом водорода одной
молекулы притягивается к атому кислорода
другой молекулы, образуя еще одну
дополнительную связь.
Водородная
связь является промежуточной между
химической и межмолекулярной. Она имеет
частично электростатический, частично
донорно-акцепторный характер.
С
повышением температуры число таких
связей сокращается. Наличие водородных
связей влияет на физические и химические
свойства веществ и объясняет высокие
температуры кипения воды и спиртов.
Здесь
в структурной формуле точками (пунктирный
штрих) показана водородная связь. Из-за
существенной разницы в электроотрицательности
водорода и кислорода(2.10 и 3.50), атом
кислорода в молекуле воды Н2О
сильно смещает к себе общие электронные
пары, в результате получается избыточный
отрицательный заряд на атоме кислорода
и положительный - на атоме водорода.
Когда две молекулы воды Н2О
оказываются рядом, атом водорода одной
молекулы притягивается к атому кислорода
другой молекулы, образуя еще одну
дополнительную связь.
Водородная
связь является промежуточной между
химической и межмолекулярной. Она имеет
частично электростатический, частично
донорно-акцепторный характер.
С
повышением температуры число таких
связей сокращается. Наличие водородных
связей влияет на физические и химические
свойства веществ и объясняет высокие
температуры кипения воды и спиртов.
|
Водородная связь осуществляется между положительно поляризованным атомом водорода одной молекулы и отрицательно поляризованным атомом другой молекулы: "Х" Н ... "Х" , где "Х" атом "F", "O", "N", реже "Cl", "S". Возникновение водородной связи обусловлено тем, что у атома водорода имеется только один электрон, который при образовании ковалентной связи с сильно электроотрицательным элементом смещается в сторону этого элемента. На атоме "Н-Н" возникает эффективный положительный заряд, что в сочетании с отсутствием внутренних электронных слоев позволяет другому атому сближаться до расстояний, близких к длинам атомных связей. Наличие водородной связи влияет на физические и химических свойства веществ. В частности, аномально высокие температуры плавления и кипения. С повышением температуры число водородных связей сокращается. |
38.
Химическое равновесие — состояние химической системы, в котором обратимо протекает одна или несколько химических реакций, причем скорости в каждой паре прямая-обратная реакция равны между собой. Для системы, находящейся в химическом равновесии, концентрации реагентов, температура и другие параметры системы не изменяются со временем.[1]
А + В ⇄ С + D
Все химические реакции, в принципе, обратимы. Это означает, что в реакционной смеси протекает как взаимодействие реагентов, так и взаимодействие продуктов. В этом смысле различие между реагентами и продуктами условное. Направление протекания химической реакции определяется условиями ее проведения (температурой, давлением, концентрацией веществ). Многие реакции имеют одно преимущественное направление и для проведения таких реакций в противоположном направлении требуются экстремальные условия. В подобных реакциях происходит почти полное превращение реагентов в продукты.
В состоянии равновесия скорости прямой и обратной реакции становятся равными.
Необратимые и обратимые реакции. Если слить растворы кислоты и щелочи, образуется соль и вода, например,
![]() ,
и если вещества были взяты в нужных
пропорциях, раствор имеет нейтральную
реакцию и в нем не остается даже следов
соляной кислоты и гидроксида натрия.
Если попытаться провести реакцию в
растворе между образовавшимися
веществами — хлоридом натрия и водой,
то никаких изменений не обнаружится. В
подобных случаях говорят, что реакция
кислоты со щелочью необратима, то есть
обратная реакция не идет. Практически
необратимы при комнатной температуре
очень многие реакции, например,
,
и если вещества были взяты в нужных
пропорциях, раствор имеет нейтральную
реакцию и в нем не остается даже следов
соляной кислоты и гидроксида натрия.
Если попытаться провести реакцию в
растворе между образовавшимися
веществами — хлоридом натрия и водой,
то никаких изменений не обнаружится. В
подобных случаях говорят, что реакция
кислоты со щелочью необратима, то есть
обратная реакция не идет. Практически
необратимы при комнатной температуре
очень многие реакции, например,
![]() ,
,
![]() и
др. Многие реакции обратимы уже в обычных
условиях, это означает, что в заметной
степени протекает обратная реакция.
Например, если попытаться нейтрализовать
щелочью водный раствор очень слабой
хлорноватистой кислоты, то окажется,
что реакция нейтрализации до конца не
идет и раствор имеет сильнощелочную
среду. Это означает, что реакция
и
др. Многие реакции обратимы уже в обычных
условиях, это означает, что в заметной
степени протекает обратная реакция.
Например, если попытаться нейтрализовать
щелочью водный раствор очень слабой
хлорноватистой кислоты, то окажется,
что реакция нейтрализации до конца не
идет и раствор имеет сильнощелочную
среду. Это означает, что реакция
![]() обратима,
то есть продукты этой реакции, реагируя
друг с другом, частично переходят в
исходные соединения. В результате
раствор имеет щелочную реакцию. Обратима
реакция образования сложных эфиров
(обратная реакция называется
омылением):RCOOH + R"OH = RCOOR" + H2O, многие
другие процессы.
обратима,
то есть продукты этой реакции, реагируя
друг с другом, частично переходят в
исходные соединения. В результате
раствор имеет щелочную реакцию. Обратима
реакция образования сложных эфиров
(обратная реакция называется
омылением):RCOOH + R"OH = RCOOR" + H2O, многие
другие процессы.
Как и многие другие понятия в химии, понятие обратимости во многом условно. Обычно необратимой считают реакцию, после завершения которой концентрации исходных веществ настолько малы, что их не удается обнаружить (конечно, это зависит от чувствительности методов анализа). При изменении внешних условий (прежде всего температуры и давления) необратимая реакция может стать обратимой и наоборот. Так, при атмосферном давлении и температурах ниже 1000° С реакцию 2 Н2 + О2 = 2 Н2О еще можно считать необратимой, тогда как при температуре 2500° С и выше вода диссоциирует на водород и кислород примерно на 4 %, а при температуре 3000° С — уже на 20 %.
В конце 19 в. немецкий физикохимик Макс Боденштейн (1871—1942) детально изучил процессы образования и термической диссоциации иодоводорода: H2 + I2 2HI. Изменяя температуру, он мог добиться преимущественного протекания только прямой или только обратной реакции, но в общем случае обе реакции шли одновременно в противоположных направлениях. Подобных примеров множество. Один из самых известных — реакция синтеза аммиака 3H2 + N2 2NH3; обратимы и многие другие реакции, например, окисление диоксида серы 2SO2 + O2 2SO3, реакции органических кислот со спиртами и т. д.
Скорость реакции и равновесие. Пусть есть обратимая реакция A + B C + D. Если предположить, что прямая и обратная реакция проходят в одну стадию, то скорости этих реакций будут прямо пропорциональны концентрациям реагентов: скорость прямой реакции v1 = k1[A][B], скорость обратной реакции v2 = k2[C][D] (квадратными скобками обозначены молярные концентрации реагентов). Видно, что по мере протекания прямой реакции концентрации исходных веществ А и В снижаются, соответственно, уменьшается и скорость прямой реакции. Скорость же обратной реакции, которая в начальный момент равна нулю (нет продуктов C и D), постепенно увеличивается. Рано или поздно наступит момент, когда скорости прямой и обратной реакций сравняются. После этого концентрации всех веществ — А, В, С и D не изменяются со временем. Это значит, что реакция достигла положения равновесия, а неизменяющиеся со временем концентрации веществ называются равновесными. Но, в отличие от механического равновесия, при котором всякое движение прекращается, при химическом равновесии обе реакции — и прямая, и обратная — продолжают идти, однако их скорости равны и поэтому кажется, что никаких изменений в системе не происходит.
Доказать протекание прямой и обратной реакций после достижения равновесия можно множеством способов. Например, если в смесь водорода, азота и аммиака, находящуюся в положении равновесия, ввести немного изотопа водорода — дейтерия D2, то чувствительный анализ сразу обнаружит присутствие атомов дейтерия в молекулах аммиака. И наоборот, если ввести в систему немного дейтерированного аммиака NH2D, то дейтерий тут же появится в исходных веществах в виде молекул HD и D2. Другой эффектный опыт был проведен на химическом факультете МГУ. Серебряную пластинку поместили в раствор нитрата серебра, при этом никаких изменений не наблюдалось. Затем в раствор ввели ничтожное количество ионов радиоактивного серебра, после чего серебряная пластинка стала радиоактивной. Эту радиоактивность не могло «смыть» ни споласкивание пластинки водой, ни промывание ее соляной кислотой. Только травление азотной кислотой или механическая обработка поверхности мелкой наждачной бумагой сделало ее неактивной. Объяснить этот эксперимент можно единственным образом: между металлом и раствором непрерывно происходит обмен атомами серебра, то есть в системе идет обратимая реакция Ag(тв) — е- = Ag+. Поэтому добавление радиоактивных ионов Ag+ к раствору приводило к их «внедрению» в пластинку в виде электронейтральных, но по-прежнему радиоактивных атомов.
Таким образом, равновесными бывают не только химические реакции между газами или растворами, но и процессы растворения металлов, осадков. Например, твердое вещество быстрее всего растворяется, если его поместить в чистый растворитель, когда система далека от равновесия, в данном случае — от насыщенного раствора. Постепенно скорость растворения снижается, и одновременно увеличивается скорость обратного процесса — перехода вещества из раствора в кристаллический осадок. Когда раствор становится насыщенным, система достигает состояния равновесия, при этом скорости растворения и кристаллизации равны, а масса осадка со временем не меняется.
Константа равновесия. Важнейший параметр, характеризующий обратимую химическую реакцию — константа равновесия К. Если записать для рассмотренной обратимой реакции A + D C + D условие равенства скоростей прямой и обратной реакции в состоянии равновесия — k1[A]равн[B]равн = k2[C]равн[D]равн, откуда [C]равн[D]равн/[A]равн[B]равн = k1/k2 = К, то величина К называется константой равновесия химической реакции.
Итак, при равновесии отношение концентрации продуктов реакции к произведению концентрации реагентов постоянно, если постоянна температура (константы скорости k1 и k2 и, следовательно, константа равновесия К зависят от температуры, но не зависят от концентрации реагентов). Если в реакции участвуют несколько молекул исходных веществ и образуется несколько молекул продукта (или продуктов), концентрации веществ в выражении для константы равновесия возводятся в степени, соответствующие их стехиометрическим коэффициентам. Так для реакции 3H2 + N2 2NH3 выражение для константы равновесия записывается в виде K = [NH3]2 равн/[H2]3равн[N2]равн. Описанный способ вывода константы равновесия, основанный на скоростях прямой и обратной реакций, в общем случае использовать нельзя, так как для сложных реакций зависимость скорости от концентрации обычно не выражается простым уравнением или вообще неизвестна. Тем не менее, в термодинамике доказывается, что конечная формула для константы равновесия оказывается верной.
Для газообразных соединений вместо концентраций при записи константы равновесия можно использовать давление; очевидно, численное значение константы при этом может измениться, если число газообразных молекул в правой и левой частях уравнения не одинаковы.
Графики, показывающие, как система приближается к равновесию (такие графики называются кинетическими кривыми), приведены на рисунках.
1. Пусть реакция необратима. Тогда k2 = 0. Примером может служить реакция водорода с бромом при 300° С. Кинетические кривые показывают изменение концентрации веществ А, B, C, D (в данном случае H2, Br2 и HBr) в зависимости от времени. Для простоты предполагается равенство исходных концентраций реагентов H2 и Br2. Видно, что концентрации исходных веществ в результате необратимой реакции снижаются до нуля, тогда как сумма концентраций продуктов достигает суммы концентраций реагентов. Видно также, что скорость реакции (крутизна кинетических кривых) максимальна в начале реакции, а после завершения реакции кинетические кривые выходят на горизонтальный участок (скорость реакции равна нулю). Для необратимых реакций константу равновесия не вводят, поскольку она не определена (К ® Ґ).
2. Пусть k2 = 0, причем k2 < k1 и К > 1 (реакция водорода с иодом при 300° С). Вначале кинетические кривые почти не отличаются от предыдущего случая, так как скорость обратной реакции мала (мала концентрация продуктов). По мере накопления HI скорость обратной реакции возрастает, а прямой — уменьшается. В какой-то момент они сравняются, после чего концентрации всех веществ уже не изменяются со временем — скорость реакции стала нулевой, хотя реакция не прошла до конца. В данном случае (K > 1) до достижения равновесия (заштрихованная часть) прямая реакция успевает пройди на значительную глубину, поэтому в равновесной смеси больше продуктов (C и D), чем исходных веществ А и В — равновесие сдвинуто вправо.
3. Для реакции этерификации уксусной кислоты (А) этанолом (В) при 50° С константа скорости прямой реакции меньше, чем обратной: k1 < k2, поэтому K < 1. Это означает, что уже при накоплении небольшого количества продуктов С и D (этилацетата и воды) скорость обратной реакции становится значительной, так что равновесие наступает, когда в смеси еще много исходных веществ. В этом случае кинетические кривые не пересекаются, а равновесие сдвинуто влево.
4. В сравнительно редком случае, когда константы скорости прямой и обратной реакций равны (k1 = k2, K = 1), для реакции A + B = C + D при [A]0 = [B]0 в равновесной смеси концентрации исходных веществ и продуктов будут одинаковыми и кинетические кривые сольются. Иногда такие условия можно создать соответствующим подбором температуры. Например, для обратимой реакции СО + Н2О = Н2 + СО2 К = 1 при температуре около 900° С. При более высоких температурах константа равновесия для этой реакции меньше 1 (например, при 1000° С К = 0,61) и равновесие сдвинуто в сторону СО и Н2О. При более низких температурах K > 1 (например, при 700° С К = 1,64) и равновесие сдвинуто в сторону СО2 и Н2.
Значение K может служить характеристикой необратимости реакции в данных условиях. Так, если K очень велика, это значит, что концентрации продуктов реакции намного превышают концентрации исходных веществ при равновесии, то есть реакция прошла почти до конца. Например, для реакции NiO + H2 Ni + H2O при 523 К (250° С) К = [Н2О]равн/[Н2]равн = 800 (концентрации твердых веществ постоянны и в выражение для К не входят). Следовательно, в замкнутом объеме после достижения равновесия концентрация паров воды будет в 800 раз больше, чем водорода (здесь концентрации можно заменить пропорциональными им давлениями). Итак, эта реакция при указанной температуре проходит почти до конца. А вот для реакции WO2 + 2H2 W + 2H2O при той же температуре К = ([Н2]равн/[Н2О]равн)2 = 10-27, следовательно, диоксид вольфрама практически не восстанавливается водородом при 500 К.
Значения К для некоторых реакций приведены в таблице.
Реакция Температура, oС К H2 + Cl2 2HCl 25 4·1031
1270 5·108
H2 + I2(г) 25 800
1035 45
I2(г) 1275 0,003
1475 0,07
3H2 + N2 25 7·105
775 0,035
СаСО3 762 100
837 300
904 800
Видно, что для одних реакций (это экзотермические реакции, идущие с выделением тепловой энергии) значение К с ростом температуры уменьшается; для других реакций (эндотермических, идущих с поглощением энергии) значение К снижается.
Константы равновесия измерены или могут быть рассчитаны для многих реакций при разных температурах, если известен тепловой эффект реакции. Количественно изменение константы равновесия с температурой определяется знаком и абсолютной величиной теплового эффекта (энтальпии) реакции DH: K = K0е-DH/RT, где K0 — постоянная, не зависящая от температуры, R — газовая постоянная, Т — абсолютная температура, е — основание натуральных логарифмов. Важнейшим успехом химической термодинамики стала возможность рассчитывать значения константы равновесия химических реакций при разных температурах и, соответственно, рассчитывать равновесные концентрации исходных веществ и продуктов без проведения многочисленных и трудоемких экспериментов. Примеры подобных расчетов.
Реакция восстановления водородом оксида железа(II) FeO + H2 Fe + H2O(г) слабо эндотермическая: DH = +23 кДж/моль (в термодинамике принято, что для экзотермических реакций DН < 0, а для эндотермических DН > 0). Для этой реакции K = [H2O]/[H2] = 0,004 при 500 К и увеличивается с повышением температуры — до 0,85 при 1500 К. Следовательно, FeO при достаточно высокой температуре восстанавливается водородом, хотя в замкнутом сосуде, если не удаляются пары воды, она идет в незначительной степени.
Реакция восстановления оксида хрома(III) Cr2O3 + 3H2 2Cr + 3H2O(г) значительно более эндотермическая: DH = +106 кДж (на 1 моль Cr2O3). Для этой реакции константа равновесия K = [H2O]3/[H2]3 = 10-23 при 500 К и даже при 1500 К она очень мала (K = 10-9). Следовательно, этот оксид не восстанавливается водородом ни при каких температурах.
Реакция восстановления оксида меди CuO + H2 Cu + H2O(г)
экзотермическая: DH = −80 кДж/моль. Константа равновесия очень велика уже при комнатной температуре (K = 1012), но скорость реакции при этом ничтожно мала. При повышении температуры эта константа уменьшается (поскольку DH < 0), но и при 500 К она все еще достаточно велика (K = 106), чтобы при нагревании реакция прошла практически до конца.
Расчеты константы равновесия очень важны для практики. Например, для синтеза аммиака увеличению К способствует понижение температуры, но чем ниже температура, тем медленнее идет реакция. Чтобы ее ускорить, нужно повышать температуру (жертвуя при этом выходом аммиака). К ускорению реакции приводит и введение катализатора. Таким образом, надо найти оптимальное для промышленного синтеза соотношение между всеми параметрами процесса, однако пока нет промышленных катализаторов, позволяющих проводить реакцию при температурах хотя бы около 100° С, когда концентрация аммиака в равновесной смеси достаточно высока, поэтому приходится использовать другой способ сдвигать равновесие в сторону аммиака — увеличивать давление, сохраняя высокую температуру.
Возникает важный для практических целей вопрос: можно ли с помощью катализатора сместить в нужную сторону химическое равновесие и таким способом увеличить выход продукта? Оказывается, нет. Введение в систему, в которой протекает обратимая реакция, катализатора приведет к снижению энергии активации как прямой, так и обратной реакции на одну и ту же величину (см. ХИМИЧЕСКАЯ КИНЕТИКА). Это означает, что катализатор в равной мере ускоряет обе реакции. Таким образом, для обратимых реакций роль катализатора заключается только в более быстром достижении равновесия. Кинетические кривые для обратимой реакции в присутствии катализатора показаны на рис. 4 пунктиром.
Равновесия в растворах электролитов: произведение растворимости. В растворах твердых электролитов (чаще всего это основания и соли) имеет место равновесие на границе двух фаз, например: AgCl(тв) = Ag+ + Cl-. Оба процесса — прямой и обратный — происходят очень быстро: достаточно в раствор с осадком AgCl добавить иодид калия и перемешать смесь, как почти сразу же весь белый хлорид серебра переходит в желтый иодид AgI; если же в раствор добавить бесцветный сульфид натрия, немедленно образуется черный сульфид серебра Ag2S.
Для подобных процессов тоже можно записать выражение константы равновесия. Эта константа носит название произведения растворимости (ПР). В общем виде для равновесия AxBy(тв) xAy+ + yBx+ ПР = [A]x[B]y. Таким образом, произведение концентраций ионов в насыщенном растворе, возведенных в соответствующие степени, представляет собой величину постоянную при данной температуре. По величине ПР ряда однотипных соединений (индексы x и y у них одинаковые) можно судить об их относительной растворимости: чем меньше ПР, тем меньше растворимость. Например, можно сравнивать растворимость (в единицах моль/л) для FeS и CuS, но нельзя сравнивать значения ПР для CuS и Ag2S (разные х). Значение ПР для некоторых малорастворимых соединений приведено в таблице.
Вещество ПР AgCl 1,8·10-10 AgBr 5,3·10-13 AgI 8,3·10-17 Ca(OH)2 6,5·10-6 Fe(OH)2 7,1·10-16 Cu(OH)2 8,3·10-20 Al(OH)3 3,2·10-34 Fe(OH)3 6,3·10-38 CaSO4 2,5·10-5 PbSO4 1,6·10-8 BaSO4 1,1·10-10 CaCO3 3,8·10-9 BaCO3 4,0·10-10 PbCO3 7,5·10-14 BaF2 1,1·10-6 CaF2 4,0·10-11 Ca3(PO4)2 2,0·10-29 Ba3(PO4)2 6,0·10-39 FeS 5·10-18 ZnS 2,5·10-22 CuS 6·10-36 Ag2S 6·10-50 HgS 1,6·10-52
Зная ПР, можно рассчитать растворимость S вещества. Так, для сульфата бария S = [Ba2+] = [SO42-] = = 1,05·10-5 моль/л, что в пересчете на массу BaSO4 составит 2,44·10-3 г/л. В случае x, y > 1 при расчетах приходится извлекать корни более высокой степени. Так, для CaF2 [Ca2+] = S, [F-] = 2S, ПР = [Ca2+][F-]2 = 4S3, для Ca3(PO4)2 ПР = (2S)2(3S)3 = 108S5 и т. д.
Поскольку при данной температуре ПР вещества постоянно, растворимость малорастворимого электролита уменьшается, если к его раствору добавить растворимый электролит, имеющий общий ион с малорастворимым электролитом. Например, если к насыщенному раствору СаСО3 (ПР = [Са2+][СО32-] добавить хорошо растворимый CaCl2, то в растворе появится очень много ионов Са2+, поэтому для постоянства ПР концентрация ионов СО32- должна сильно уменьшиться и равновесие СаСО3(тв) Ca2+ + CO32- сдвинется влево с образованием дополнительного количества осадка. То же произойдет, если к раствору добавить Na2CO3.
Равновесия в растворах электролитов: константа диссоциации. Количественно силу кислот в водных растворах характеризует константа диссоциации кислоты Кa (от англ. acid — кислота), которая определяется как константа равновесия процесса диссоциации. Рассмотрим реакцию диссоциации уксусной кислоты: СН3СООН СН3СОО- + H+. Пусть с0 — исходная концентрация кислоты; очевидно, что при равновесии [СН3СООН]равн = с0 — [H+]равн и [СН3СОО-]равн = [H+]равн. Тогда для константы равновесия можно записать уравнение K = [СН3СОО-]равн[H+]равн/(с0 — [H+]равн) = [H+]2равн/(с0 — [H+]равн). Решая это квадратное уравнение (и опуская для простоты обозначение «равн» и индекс «а» для К), получаем: [H+] = (-K +)/2 (очевидно, что знак минус перед корнем не имеет физического смысла).
Из выражения для Кa видно, что для сильных кислот, которые диссоциируют почти полностью, концентрации ионов Н+ и А- будут близки к исходной концентрации кислоты, введенной в раствор, тогда как концентрация недиссоциированных молекул НА в растворе будет близка к нулю. В результате константа диссоциации будет очень большой. С другой стороны, для слабых кислот константа будет мала. Например, для уксусной кислоты Кa = 1,8·10-5, а для более сильной муравьиной Кa = 1,8·10-4. Для самых сильных кислот значение Кa может превышать 1010.
Если кислота диссоциирует в малой степени, выведенная формула для расчета концентрации ионов водорода заметно упрощается. Действительно, при слабой диссоциации концентрация молекул кислоты с0 практически не уменьшается, так что с0 " [H+]. При этом выражение для константы равновесия запишется в виде К = [H+]2/с0, откуда [H+] = . Уксусная кислота относится к слабым кислотам; при комнатной температуре значение К для нее равно 1,75·10-5, то есть равновесие сдвинуто влево — в сторону недиссоциированных молекул кислоты. При с0 = 1 моль/л [H+] = = 0,0042 моль/л, то есть продиссоциировала очень малая часть молекул. Степень диссоциации (ее можно рассчитать по формуле) в данном случае равна 0,0042 или 0,42 %. С разбавлением кислоты степень диссоциации увеличивается. Например, при c0 = 0,01 моль/л [H+] = = 0,00042 моль/л, а степень диссоциации равна = 0,042 или 4,2 %. Если кислота многоосновная (например, Н3РО4), то для нее приходится вводить несколько констант диссоциации, соответствующих отщеплению одного, двух и т. д. ионов Н+. Так, для фосфорной кислоты Кa = 7,5·10-3 при диссоциации по первой ступени, 6,3·10-8 — по второй ступени и 1,3·10-12 — по третьей.
Для удобства вместо константы диссоциации обычно пользуются величиной рКa, которая называется показателем кислотности и определяется (по аналогии с водородным показателем) выражением рКа = -lgКa. Здесь чем меньше величина рКa, тем кислота сильнее. Так, для уксусной кислоты рКa = 4,7, для муравьиной рКa = 3,7. Для большинства известных кислот рКa принимает значения в интервале от 1 до 14. Самые сильные кислоты, диссоциирующие в водном растворе практически нацело, имеют рКa < 0.
Значения рКa для некоторых кислот при комнатной температуре приведены в таблице (для многоосновных кислот приведено значение, характеризующее диссоциацию только по первой ступени). Кислоты приведены в порядке уменьшения величины рКа, то есть в порядке увеличения силы кислоты.
Кислота pКa H2O2 11,6 C6H5OH 10,0 H2SiO3 9,7 H2SnO3 9,4 H3BO3 9,2 HCN 9,1 H2S 7,2 H2CO3 6,4 CH3COOH 4,8 C6H8O6(аскорбиновая) 4,1 НСООН 3,8 HNO2 3,4 HF 3,2 C6H8O7(лимонная) 3,1 С4Н6О6(винная) 3,0 H3PO4 2,1 H2SO3 1,8 CCl3COOH 1,7 Н2С2О4 1,3 HIO3 0,8 H2CrO4 −1 HNO3 −1,6 HMnO4 −2,3 H2SO4 −3 HCl −7 HBr −9 HI −11
Аналогично вводится понятие для константы диссоциации оснований и соответствующего показателя основности pKb (от англ. base — основание); оно используются главным образом для слабых органических оснований, которые при растворении в воде дают щелочную среду: B + H2O BH+ + OH-. Так, для анилина C6H5NH2 (слабое основание) рКb = 9,4, а для гуанидина HN=C(NH2)2 (сильное основание) рКb = 0,55.
Для воды, которая является одновременно слабой кислотой и основанием равной силы: Н2О Н+ + ОН- также можно записать константу равновесия К = [H+][OH-]/[H2O]. В чистой воде концентрация [Н2О] — величина постоянная, равная 55,6 моль/л. Это значение мало изменяется и в разбавленных водных растворах кислот и оснований. Поэтому для таких растворов произведение [H+][OH-] = К[H2O] является также величиной постоянной и называется ионным произведением воды. При 25° С оно равно 10-14, и, соответственно, [Н+] = [ОН-] = 10-7 моль/л (см. ВОДОРОДНЫЙ ПОКАЗАТЕЛЬ (PН)).
Принцип Ле Шателье. В 1884 французский физикохимик и металлург Анри Луи Ле Шателье (1850—1936) сформулировал общий закон смещения химического равновесия: «Если на систему, находящуюся в состоянии химического равновесия, оказывать внешнее воздействие (изменять температуру, давление, концентрации веществ), то положение равновесия смещается в такую сторону, чтобы ослабить внешнее воздействие». В любом случае равновесие будет смещаться до тех пор, пока не наступит новое положение равновесия, которое соответствует новым условиям. Этот принцип позволяет легко предсказать качественные изменения в равновесной системе при изменении условий.
Как система может «противодействовать» изменению внешних условий? Если, например, температуру равновесной смеси повышают нагреванием, сама система, конечно, не может «ослабить» внешний нагрев, однако равновесие в ней смещается таким образом, что для нагревания реакционной системы до определенной температуры требуется уже большее количество теплоты, чем в том случае, если бы равновесие не смещалось. При этом равновесие смещается так, чтобы теплота поглощалась, то есть в сторону эндотермической реакции. Это и можно трактовать как «стремление системы ослабить внешнее воздействие». С другой стороны, если в левой и правой частях уравнения имеется неодинаковое число газообразных молекул, то равновесие в такой системе можно сместить и путем изменения давления. При повышении давления равновесие смещается в ту сторону, где число газообразных молекул меньше (и таким способом как бы «противодействует» внешнему давлению). Если же число газообразных молекул в ходе реакции не меняется
(H2 + Br2(г) 2HBr, СО + Н2О(г) СО2 + Н2), то давление не влияет на положение равновесия. Следует отметить, что при изменении температуры изменяется и константа равновесия реакции, тогда как при изменении только давления она остается постоянной.
Несколько примеров использования принципа Ле Шателье для предсказания смещения химического равновесия. Реакция 2SO2 + O2 2SO3(г) экзотермична. Если повысить температуру, преимущество получит эндотермическая реакция разложения SО3 и равновесие сместится влево. Если же понизить температуру, равновесие сместится вправо. Так, смесь SО2 и О2, взятых в стехиометрическом соотношении 2:1 (см. СТЕХИОМЕРИЯ), при температуре 400° С и атмосферном давлении превращается в SО3 с выходом около 95 %, то есть состояние равновесия в этих условиях почти полностью смещено в сторону SО3. При 600° С равновесная смесь содержит уже 76 % SО3, а при 800° С — только 25 %. Именно поэтому при сжигании серы на воздухе образуется в основном SО2 и лишь около 4 % SО3. Из уравнения реакции следует также, что повышение общего давления в системе будет сдвигать равновесие вправо, а при понижении давления равновесие будет смещаться влево.
Реакцию отщепления водорода от циклогексана с образованием бензола
С6Н12 С6Н6 + 3Н2 проводят газовой фазе, также в присутствии катализатора. Реакция эта идет с затратой энергии (эндотермическая), но с увеличением числа молекул. Поэтому влияние температуры и давления на нее будет прямо противоположным тому, которое наблюдается в случае синтеза аммиака. А именно: увеличению равновесной концентрации бензола в смеси способствует повышение температуры и понижение давления, поэтому реакцию проводят в промышленности при невысоких давлениях (2-3 атм) и высоких температурах (450-500° С). Здесь повышение температуры «дважды благоприятно»: оно не только увеличивает скорость реакции, но и способствует сдвигу равновесия в сторону образования целевого продукта. Конечно, еще большее снижение давления (например, до 0,1 атм) вызвало бы дальнейшее смещение равновесия вправо, однако при этом в реакторе будет находиться слишком мало вещества, уменьшится и скорость реакции, так что общая производительность не повысится, а понизится. Этот пример еще раз показывает, что экономически обоснованный промышленный синтез — это удачное лавирование между «Сциллой и Харибдой».
Принцип Ле Шателье «работает» и в так называемом галогенном цикле, который используют для получения титана, никеля, гафния, ванадия, ниобия, тантала и других металлов высокой чистоты. Реакция металла с галогеном, например, Ti + 2I2 TiI4 идет с выделением теплоты и потому при повышение температуры равновесие смещается влево. Так, при 600° С титан легко образует летучий иодид (равновесие смещено вправо), а при 110° С иодид распадается (равновесие смещено влево) с выделением очень чистого металла. Такой цикл работает и в галогенных лампах, где испарившийся со спирали и осевший на более холодных стенках вольфрам образует с галогенами летучие соединения, которые на раскаленной спирали вновь распадаются, и вольфрам оказывается перенесенным на прежнее место.
Кроме изменения температуры и давления существует еще один действенный способ влиять на положение равновесия. Представим, что из равновесной смеси
А + В C + D выводится какое-либо вещество. В соответствии с принципом Ле Шателье, система тут же «отзовется» на такое воздействие: равновесие начнет смещаться так, чтобы скомпенсировать потерю данного вещества. Например, если из зоны реакции выводить вещество С или D (или оба сразу), равновесие будет смещаться вправо, а если выводить вещества А или В — влево. Введение какого-либо вещества в систему также будет смещать равновесие, но уже в другую сторону.
Удалять вещества из зоны реакции можно разными способами. Например, если в плотно закрытом сосуде с водой есть сернистый газ, установится равновесие между газообразным, растворенным и прореагировавшим диоксидом серы:
О2(г) SО2(р) + Н2О H2SO3. Если сосуд открыть, сернистый газ постепенно начнет улетучиваться и больше не сможет участвовать в процессе — равновесие начнет смещаться влево, вплоть до полного разложения сернистой кислоты. Аналогичный процесс можно наблюдать каждый раз при открывании бутылки с лимонадом или минеральной водой: равновесие СО2(г) СО2(р) + Н2О Н2СО3 по мере улетучивания СО2 смещается влево.
Вывод реагента из системы возможен не только при образовании газообразных веществ, но и путем связывания того или иного реагента с образованием нерастворимого соединения, выпадающего в осадок. Например, если в водный раствор СО2 ввести избыток соли кальция, то ионы Са2+ будут образовывать осадок СаСО3, реагируя с угольной кислотой; равновесие СО2(р) + Н2О Н2СО3 будет смещаться вправо, пока в воде не останется растворенного газа.
Равновесие можно сместить и добавлением реагента. Так, при сливании разбавленных растворов FeCl3 и KSCN появляется красновато-оранжевая окраска в результате образования тиоцианата (роданида) железа:
FeCl3 + 3KSCN Fe(SCN)3 + 3KCl. Если в раствор внести дополнительно FeCl3 или KSCN, окраска раствора усилится, что свидетельствует о смещении равновесия вправо (как бы ослабляя внешнее воздействие). Если же добавить к раствору избыток KCl, то равновесие сместится влево с ослаблением окраски до светло-желтой.
В формулировке принципа Ле Шателье недаром указывается, что предсказывать результаты внешнего воздействия можно только для систем, находящихся в состоянии равновесия. Если этим указанием пренебречь, легко прийти к совершенно неверным выводам. Например, известно, что твердые щелочи (KOH, NaOH) растворяются в воде с выделением большого количества теплоты — раствор разогревается почти так же сильно, как и при смешении с водой концентрированной серной кислоты. Если забыть, что принцип применим только к равновесным системам, можно сделать неверный вывод о том, что при повышении температуры растворимость КОН в воде должна снижаться, так как именно такое смещение равновесия между осадком и насыщенным раствором приводит к «ослаблению внешнего воздействия». Однако процесс растворения КОН в воде — вовсе не равновесный, поскольку в нем участвует безводная щелочь, тогда как осадок, находящийся в равновесии с насыщенным раствором, представляет собой гидраты КОН (в основном KOH·2H2O). Переход же этого гидрата из осадка в раствор является эндотермическим процессом, то есть сопровождается не нагреванием, а охлаждением раствора, так что принцип Ле Шателье для равновесного процесса выполняется и в этом случае. Точно так же при растворении безводных солей — CaCl2, CuSO4 и др. в воде раствор нагревается, а при растворении кристаллогидратов CuSO4·5H2O, CaCl2·6H2O — охлаждается.
В учебниках и популярной литературе можно найти еще один интересный и поучительный пример ошибочного использования принципа Ле Шателье. Если в прозрачный газовый шприц поместить равновесную смесь бурого диоксида азота NO2 и бесцветного тетраоксида N2O4, а потом с помощью поршня быстро сжать газ, то интенсивность окраски сразу же усилится, а через некоторое время (десятки секунд) вновь ослабится, хотя и не достигнет первоначальной. Этот опыт обычно объясняют так. Быстрое сжатие смеси приводит к увеличению давления и, следовательно, концентрации обоих компонентов, поэтому смесь становится более темной. Но повышение давления, в соответствии с принципом Ле Шателье, сдвигает равновесие в системе 2NO2 N2O4 в сторону бесцветного N2O4 (уменьшается число молекул), поэтому смесь постепенно светлеет, приближаясь к новому положению равновесия, которое соответствует повышенному давлению.
Ошибочность такого объяснения следует из того, что обе реакции — диссоциация N2O4 и димеризация NO2 — происходят чрезвычайно быстро, так что равновесие в любом случае устанавливается за миллионные доли секунды, поэтому невозможно вдвинуть поршень настолько быстро, чтобы нарушить равновесие. Объясняется этот опыт иначе: сжатие газа вызывает значительно повышение температуры (с этим явлением знаком каждый, кому приходилось накачивать шину велосипедным насосом). И в соответствии с тем же принципом Ле Шателье, равновесие мгновенно сдвигается в сторону эндотермической реакции, идущей с поглощением теплоты, то есть в сторону диссоциации N2O4 — смесь темнеет. Затем газы в шприце медленно остывают до комнатной температуры и равновесие снова сдвигается в сторону тетраоксида — смесь светлеет.
Принцип Ле Шателье прекрасно действует и в тех случаях, которые не имеют никакого отношения к химии. В нормально действующей экономике общая сумма находящихся в обращении денег находится в равновесии с теми товарами, которые можно на эти деньги купить. Что будет, если «внешним воздействием» окажется желание правительства напечатать денег побольше, чтобы рассчитаться с долгами? В строгом соответствии с принципом Ле Шателье, равновесие между товаром и деньгами будет смещаться таким образом, чтобы ослабить удовольствие граждан от обладания большим количеством денег. А именно, цены на товары и услуги вырастут, и таким путем будет достигнуто новое равновесие. Другой пример. В одном из городов США было решено избавиться от постоянных пробок путем расширения магистралей и строительства транспортных развязок. На некоторое время это помогло, но затем обрадованные жители начали покупать больше автомобилей, так что вскоре пробки возникли вновь — но при новом «положении равновесия» между дорогами и бóльшим числом автомобилей.
Равновесие в окислительно-восстановительных реакциях в растворе. Для того, чтобы определить термодинамическую возможность протекания окислительно-восстановительных реакций в водных растворах, используют понятие электрического потенциала Е, выраженного в вольтах (см. ХИМИЧЕСКИЕ ИСТОЧНИКИ ТОКА). Для реакции aA + bB cC + dD в качестве критерия используют уравнение Нернста:
Е = Е0 — (RT/nF)ln([C]c[D]d/[A]a[B]b), где Е0 — стандартный потенциал, n — число переданных от окислителя к восстановителю электронов, F — постоянная Фарадея. Если ограничиться температурой 25° С (Т = 298 К), подставить значения R и F, и перейти от натуральных логарифмов к десятичным, то получим: E = E0 -(0,058/n)lg([C]c[D]d/[A]a[B]b). Если для данной реакции рассчитанное значение Е > 0, то реакция не пойдет, вернее, равновесие будет смещено влево и тем сильнее, чем больше Е. Если Е < 0, равновесие смещено вправо и тем сильнее, чем более отрицательно Е.
Используя уравнение Нернста, можно рассмотреть такую «странную» реакцию:
Cu + 2H+ = Cu2+ + H2 и выяснить, каков будет ее потенциал Е в разных условиях. Пусть, для простоты, давление водорода равно 1 атм, а концентрация ионов водорода в растворе равна 1 моль/л, то есть меняется только концентрацию ионов меди. Учитывая, что все окислительно-восстановительные потенциалы принято записывать как реакции восстановления (например, для меди Cu2+ + 2e ® Cu), уравнение Нернста для рассматриваемой реакции получается в виде: E = E0 + 0,029lg[Cu2+].
Пусть в растворе уже есть соль меди, причем [Cu2+] = 1 моль/л. Тогда lg[Cu2+] = 0 и E = E0. Стандартный электродный потенциал для меди Ео известен и равен +0,34 В. Это значит, что в рассмотренных условиях медь не будет переходить в раствор в виде ионов. Напротив, термодинамически разрешена обратная реакция Cu2+ + H2 = Cu + 2H+; возможность вытеснения металлов (меди, серебра, ртути) из растворов их солей водородом (под давлением) доказал в 1865 русский химик Н. Н. Бекетов. А можно ли заставить реакцию идти в обратном направлении, то есть растворить медь в кислоте? Для сдвига равновесия вправо нужно снизить значение Е и сделать его отрицательным. Как видно из формулы Нернста, для уменьшения Е надо уменьшить концентрацию ионов меди в растворе. Формула показывает также, что это снижение должно быть очень сильным. Действительно, даже если в растворе будет всего 10-9 моль/л ионов меди, то есть в миллиард раз меньше стандартной концентрации, потенциал
Е = E0 + 0,029lg[Cu2+] = 0,34 + 0,029lg(10-9) = 0,034 + 0,029(-9) = +0,08 B и реакция не пойдет. А ведь при такой ничтожной концентрации в 1 л раствора содержится всего лишь 0,000064 мг ионов меди. Даже в чистой воде растворимость меди выше, не говоря уже о соляной или серной кислотах. Потому-то медь не растворяется ни в воде, ни в разбавленных кислотах. Вернее, растворение идет только первые мгновения, пока меди в растворе совсем нет и Е < 0, но как только концентрация ионов меди достигнет ничтожно малого значения, потенциал Е станет положительным и реакция остановится (речь здесь не идет о кислотах-окислителях типа азотной или горячей концентрированной серной кислоты, в которых механизм растворения меди совсем другой.)
Легко подсчитать, при каких концентрациях ионов меди потенциал Е снизится до нуля и далее станет отрицательным. Условию Е = 0 соответствует уравнение
0,34 + 0,029lg[Cu2+] = 0, откуда lg[Cu2+] = −11,7 или приближенно [Cu2+] = 10-12 моль/л. Можно ли практически достичь таких малых значений? Химикам известны различные способы снижения концентрации ионов меди (и других металлов) до очень низких значений. Один из них — связывание ионов в очень прочные комплексы, которые почти не диссоциируют с образованием свободных ионов. К таким мощным комплексообразователям относятся, например, цианид-ионы. Поэтому в присутствии цианида калия медь будет растворяться даже в воде (при этом идет только одноэлектронное окисление до Cu+):
2Cu + H2O + 4KCN = 2K[Cu(CN)2] + 2KOH + H2.
Дополнительный фактор, который способствующий снижению потенциала Е — высокая концентрация ионов водорода в растворе. Но практически повысить эту концентрацию можно не очень сильно; например, в концентрированной соляной кислоте [H+] = 10 моль/л. Но в этих условиях анионы Cl-, которых в растворе тоже много, способны связывать в довольно прочные комплексы однозарядные катионы Cu+. В результате медь может медленно реагировать и с концентрированной соляной кислотой: Cu + 4HCl = 2H[CuCl2] + H2 (окислению меди способствует и растворенный кислород). В растворах же крепкой иодоводородной кислоты растворяется с выделением водорода даже серебро, поскольку ионы Ag+ в этом случае связываются в очень прочные иодидные комплексы, почти не диссоциирующие на ионы I- и Ag+.
40.
Комплексные соединения (лат. complexus — сочетание, обхват) или, другими словами, координационные соединения — это частицы (нейтральные молекулы или ионы), которые образуются в результате присоединения к данному иону (или атому), называемому комплексообразователем (центральным атомом или металлоцентром; в современной научной литературе доминирует термин «металлоцентр»), нейтральных молекул или других ионов, называемых лигандами.
Комплексные соединения мало диссоциируют в растворе (в отличие от двойных солей). Комплексные соединения могут содержать комплексный малодиссоциирующий анион ([Fe(CN)6]3−), комплексный катион ([Ag(NH3)2]+) либо вообще не диссоциировать на ионы (соединения типа неэлектролитов, например карбонилы металлов). Комплексные соединения разнообразны и многочисленны.
Применяются в химическом анализе, в технологии при получении ряда металлов (золота, серебра, металлов платиновой группы и др.), для разделения смесей элементов, например, лантаноидов.
Огромная область применения комплексов переходных металлов — каталитические процессы.