

Электропроводность электролитов.
Закон Ома: I = U/R. При заданной длине проводника l и площади его поперечного сечения S сопротивление R определяется удельной электропроводностью κ или удельным сопротивлением ρ материала проводника:
κ = 1/ ρ = l / (S R), (Ом ∙м)-1
Ионная проводимость растворов электролитов (проводники 2-го рода) обусловлена их диссоциацией Кν+А ν- = ν+ Кz+ + ν− Az-, ν+ z+ = ν− z – .
Скорость миграции ионов под действием поля напряженностью Е:
![]() определяется
балансом действующих на ион сил
электрического поля и трения Стокса (
z
e
E
=
6 π
η
r
υ
).
определяется
балансом действующих на ион сил
электрического поля и трения Стокса (
z
e
E
=
6 π
η
r
υ
).
Подвижность ионов – это умноженная на число Фарадея удельная скорость λ± = F υ ± /E = F z±e / (6 π η r±), (5.27)
где η – вязкость среды, r± - эффективный радиус ионов.
При бесконечном разбавлении справедлив закон независимого движения ионов (Кольрауш): λ∞ = λ∞+ +λ∞− При этом λ∞ η = const – Правило Вальдена).
В общем случае эквивалентная электропроводность пропорциональна степени диссоциации α λ = κ∙1000/(С, моль/л) = λ∞ ∙α ∙fλ ,
где fλ - коэффициент электропроводности – аналог коэффициента активности, характеризующий межионное взаимодействие.
Для слабых электролитов f λ ≈ 1 и решение для α из закона разведения Оствальда при концентрациях С << Кд может быть приближенно представлено в виде α ≈ 1 – С/ Кд . При этом λ = λ∞(1- С/ Кд).
Для разбавленных растворов сильных электролитов (α ≈ 1) Кольрауш экспериментально нашел, а Онсагер теоретически обосновал концентрационную зависимость "квадратного корня". λ = λ∞∙f λ = λ∞ − const √С.
Вышеприведенные зависимости из измерений электропроводности позволяют определять эффективные размеры ионов (r+ , r−) , вязкость среды, наконец количественные закономерности в поведении степени диссоциации электролитов. Измерение электрической проводимости электролитов – кондуктометрия - кроме определения физико-химических характеристик применяется в химическом анализе (кондуктометрическое титрование по минимуму электропроводности в точке эквивалентности).
В проводимости одновременно участвуют анионы и катионы. Их доли в эквивалентной электропроводности обычно неодинаковы и характеризуются числами переноса. t± = λ ± / λ = v ± / v .
Числа переноса определяются либо прямым измерением скоростей движения окрашенных ионов в электрическом поле, либо по изменению концентраций в прианодном и прикатодном пространствах до и по окончании электролиза (Гитторф)
5.9. Законы Фарадея.
где I – ток, z - электрохимическая валентность.
Объединенная форма раскрывает физический смысл эмпирически установленных законов Фарадея.
Масса m вещества, претерпевшего химическое превращение, пропорциональна электрическому заряду q, прошедшему через электролит. Коэффициент пропорциональности – Кэ = Мэ / F- электрохимический эквивалент.
Электрохимические эквиваленты разных веществ пропорциональны их химическим эквивалентам. Коэффициент пропорциональности 1/F определяет число Фарадея, заряд иона zF / NA и его кратность элементарному заряду e.
Законы Фарадея являются общими и точными. Отклонения обусловлены побочными неэлектрохимическими составляющими. При этом эффективность или КПД собственно электрохимического процесса оценивается выходом по току. Ниже представлен отсутствующий в литературе вывод закона Фарадея.
Закон кратных отношений в соответствии со стехиометрией химической реакции 0 = Σ νi Ai , (νi < 0 для реагентов) определяет изменение количества вещества всех участников реакции через единую реакционную переменную – координату реакции ξ: (ni - n0i)/νi = Δni /νi = ξ.
Его применение к
электродной полуреакции Mz+
+ z e–
= M
(0 = - Mz+
- z e–
+ M),
с одной стороны, к электронам -Δnе
/(-z)=
ξ, с другой - , к металлу ΔnM
=
ξ, формирует равенство: ΔnM
=
Δnе
/z,
из которого после очевидных подстановок
ΔnM
=
m/M
и
Δnе
= q/F
следует
объединенный закон Фарадея
![]() .
.
5.10. Скорость электрохимической реакции
как гетерогенной реакции определяется изменением числа молей n к единице поверхности электрода S (см. раздел "Химическая кинетика").
υ = (1/S) dn / dt.
При использовании законов Фарадея эта скорость может быть выражена через плотность тока i = I / S . (n =m /M = i S t / zF)
υ = i / zF.
Эксплуатация химического источника тока или электролиз (см. далее) является неравновесными, чаще стационарными процессами и характеризуется величиной электродного потенциала φ отличающегося от равновесного φр. Их разность, как мы увидим далее, пропорциональная току, называется электродной поляризацией η = φ – φр.
В отношении отдельных составляющих (стадий) скорости гетерогенной электрохимической реакции используют термин перенапряжение.
Первая стадия – транспорт реагентов к электроду и продуктов от электрода в электролит.
Вторая, собственно электрохимическая стадия, связана с переходом электронов и ионов через границу раздела раствор – электрод. Эту стадию обычно сопровождают и дополняют предшествующие или последующие реакции а) химических и б) фазовых превращений.
5.11. Транспортное, диффузионное перенапряжение
Транспорт участников реакции осуществляется за счет
миграции заряженных частиц, взаимосвязанной с числами переноса;
диффузии в приэлектродный слой с необходимостью учета конвективной, потоковой ее составляющей.
Для простейшей электрохимической системы под током рассмотрим случай, когда транспорт ионов будет более медленной стадией по сравнению с электрохимической.
+ M │ Ca Mz+(C) Ck│ M –
Скорость установившегося, стационарного прикатодного процесса vк = vмигр + vдифф с учетом электрохимического её определения и конкретизацией диффузионной составляющей равна
i / (zF) = tк i / (zF) + D (C - Ck) / δ ,
где tk – число переноса для катионов; D - коэффициент диффузии ионов, δ – эффективная толщина диффузионного слоя.
|
|
Концентрация катионов в обедненном прикатодном слое Ck, которая и определяет неравновесный электродный потенциал под током, равна Ck = C (1 – i /(z iпр)),
где iпр = DFC/(δ (1 – tк)), - предельный ток. |
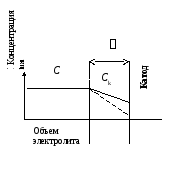
После подстановки Ck в ηк получаем искомое диффузионное перенапряжение катодного процесса как функцию тока
ηк = φ – φравн = φº + (RT/(zF)) lnCк – [φº +(RT/(zF)) lnC ]
ηк,а = (RT/(zF)) ln(1 – i /(z iпр)),
Для удобства графического отображения полученных зависимостей η(i) и обратной ей i(η) условимся внутри электрохимической ячейки считать ток, входящий в электрод – положительным, а выходящий – отрицательным. Последняя формула с учетом знака z принимает единый вид и для катода и для анода, а за ней следует обратная зависимость тока на электродах от перенапряжения.
η = (RT/(zF)) ln(1 – i / (z iпр)), i = z iпр (1 – exp (z y)),
где y = η RT/F – безразмерное перенапряжение. На катоде y, η < 0, i → z iпр при η → - ∞. На аноде y, η > 0, а i → ∞ при увеличении η.
|
| ||
|
Рассмотренная модель диффузионного перенапряжения как для катодного, так и для анодного процессов учитывает миграцию заряженных частиц и их диффузию, в том числе конвективную.
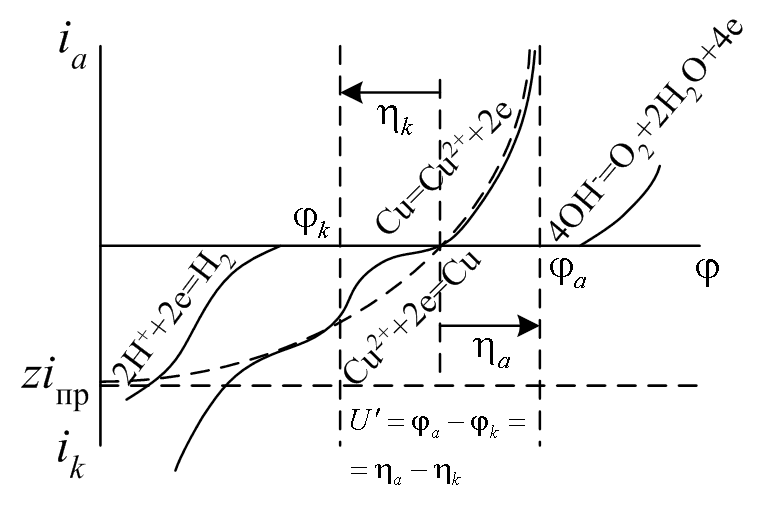
Химическую составляющую (химическое перенапряжение) электродного процесса упрощенно представляют в виде промежуточных химических реакций, дополняющих собственно электродную Ox + z e– Red.
Например, собственно электрохимической стадии катодного выделения меди (реакция (b)) из раствора Cu(CN)2– предшествует реакция (a) образования окислителя (Ox =CuCN)
Cu(CN)2– CuCN + CN– (a) CuCN + e– Cu + CN– (b).
Аналогичным образом в виде квазихимических реакций можно выразить и фазовое перенапряжение, т.е. торможение электродной реакции за счет сопутствующих фазовых превращений (образование газов, покрытий и т.д.). Например, для катодной электрокристаллизации металлов дополнительно учитывается стадия зародышеобразования.
В практических исследованиях перенапряжение делят на диффузионное и активационное. При такой расширенной трактовке выражения для катодного и анодного перенапряжений включают в себя соответствующие кинетические параметры.
![]() ,
, ![]() ,
,
где i0 - плотность тока обмена, кроме диффузионных, включает в себя и кинетические (активационные) характеристики гетерогенных электродных процессов.
В области малых перенапряжений |zy| << 1, exp (zy) ≈ 1+ zy
![]() ,
,
![]() .
.
Вывод: при изготовлении электродов сравнения, обеспечивающих постоянство равновесного потенциала, используются системы с большим током обмена i0.
В области больших перенапряжений z y >> 1
![]()
![]()
эмпирическое уравнение Тафеля используется, в частности, для характеристики электролитического выделения водорода. Например, для кислых растворов (H3O+ + e– 1/2 H2 + H2O)
|
Металл |
Pt |
Au |
Ni |
Fe |
Cu |
Ag |
Sn |
Zn |
Hg |
Pb |
|
a, В |
0,1 |
0,4 |
0,63 |
0,7 |
0,87 |
0,95 |
1,2 |
1,24 |
1,41 |
1,56 |
|
b, В |
0,03 |
0,12 |
0,11 |
0,12 |
0,12 |
0,1 |
0,13 |
0,12 |
0,11 |
0,11 |
На величину поляризации существенно влияет состояние поверхности: ее уменьшает шероховатость; покрытие платиновой черни на поверхности платины снижает ее до нуля. Перенапряжение водорода падает с повышением температуры (за счет активационного увеличения i0).
Кстати, расчетное значение таффелевского коэффициента b = 0,118 В при β = 0,5 и Т = 298 К в большинстве случаев совпадает с наблюдаемыми величинами.
Механизм и последовательная теоретическая модель электродной поляризации до сих пор не установлены. На качественном уровне эксперимент интерпретируется совокупностью последовательных стадий.
1) массодоставка H+ (H3O+) из раствора к электроду;
2) адсорбция, дегидратация и разряд H+;
3)
образование молекулярного водорода: ;
;
4) десорбция с пузырьковым фазо-, точнее, газообразованием.
В настоящее время лимитирующей и определяющей считается вторая стадия "замедленного разряда". На величину перенапряжения водорода влияют состав раствора, pH, общая концентрация ионов, наличие ПАВ и т.д.
Величина
водородного перенапряжения, нежелательная
с точки зрения получения водорода, может
играть положительную роль, например, в
технологии электролиза водных растворов
солей цинка. Вследствие высокого
перенапряжения водорода на Zn потенциал
его выделения (в нейтральном растворе
![]() =
–0,41 В) сдвигается в сторону отрицательных
значений, благодаря чему возможно
катодное осаждение металла (
=
–0,41 В) сдвигается в сторону отрицательных
значений, благодаря чему возможно
катодное осаждение металла (![]() =
–0,76 В) с выходом по току до 95 %.
=
–0,76 В) с выходом по току до 95 %.
Режим электролиза для гальванического элемента – это принудительная реализация невыгодной восстановительной полуреакции путем создания избытка электронов, а значит отрицательного потенциала на катоде. Положительный потенциал на аноде от внешнего источника призван нейтрализовать электроны также невыгодной окислительной полуреакции.
Главное качество и назначение электролиза заключается в проведении ОВР, в более широком смысле процессов, самопроизвольное протекание которых по термодинамике невозможно. Например, разложение одномолярного раствора HCl (1 М) на элементы сопровождается возрастанием энергии Гиббса на 131,26 кДж/моль:
1/2 Cl2 + e– Cl– L = 1,36 В
H+ + e– 1/2 H2 R = 0
H+ + Cl– = 1/2 H2 + 1/2 Cl2 , E = –1,36 В G = –FE = 131,26 кДж/моль
Для осуществления электролиза, в соответствии с рис. 5.12. (I) к электролизёру необходимо приложить разность потенциалов или напряжение
U = U′ + U = a – k + U ,
которое кроме омического падения напряжения электролита, электродов и контактов U, включает в себя разность потенциалов поляризованных электродов под током U′ = a – k , называемую напряжением разложения электролита:
U′ = p,a + |a| – (p,k – |k|) = p,a – p,k + |a| + |k|= U0 + |a| +|k|.
Здесь указанные электродные потенциалы выражены через соответствующие равновесные потенциалы и электродные поляризации (.
|
|
|
Рис. 5.12. Катодные (1) и анодные (2) поляризационные кривые при электролизе (I) и работе гальванического элемента (II) |
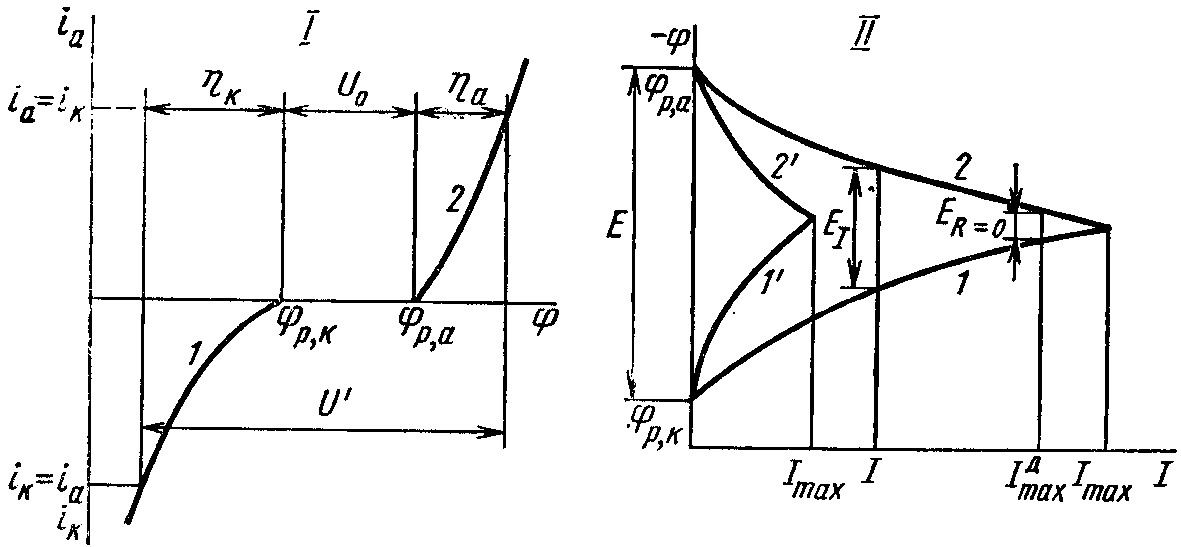
При
этом разность равновесных электродных
потенциалов называется обратимым
напряжением разложения электролита:
U0
= p,a
– p,k
, которое численно равно э.д.с.
электрохимической ячейки в режиме
гальванического элемента, и которое
соответствует реакции, обратной реакции
при электролизе. Например,
![]() , T =
298,15 К.
, T =
298,15 К.
Напряжение разложения воды на платиновых электродах с учетом перенапряжений на электродах составляет U' = 1,7 В. Эта же величина соответствует и кислородсодержащим кислотам и щелочам, поскольку при электролизе их водных растворов идет разложение воды.
Процессы, протекающие при электролизе, можно разбить на три группы:
1) При катодном осаждении металла с использованием растворимого анода из того же металла в случае равенства катодного и анодного выхода по току электродные реакции обратны друг другу и химических превращений в электролизере не происходит (пример электролиза CuSO4 с медными элктродами).
2) Электролиз раствора соляной кислоты - пример электролиза сопровождающегося химическим разложением электролита.:
3) Химические реакции, в которых участвуют различные компоненты электролита и растворитель.
Примером может служить электролиз кислого раствора KCl с инертным анодом:
|
Возможные реакции на аноде |
, В |
Возможные реакции на катоде |
, В |
|
А1: 1/2 Cl2 + e– Clˉ |
1,36 |
К1: К+ + e- К |
-2,93 |
|
А2: ¼ O2 + H+ + e- 1/2 H2O |
1,23 |
К2: H+ + e– 1/2 H2 |
0 |
Выбор полуреакций по минимальной катодно-анодной разности потенциалов, соответствующей минимуму энергетических затрат, дает реакции А2 и К2, однако по меньшему перенапряжению вместо кинетически заторможенной реакции А2 с выделением кислорода следует выбрать А1 , с выделением хлора. Т.о. результирующая реакция соответствует сумме подчеркнутых: H+ + Clˉ = 1/2 H2 + 1/2 Cl2
Широкому практическому применению электролиза способствует высокое качество продуктов (например, чистота) и достаточная экономич-ность метода. Электролиз является практически единственным способом получения важнейших металлов, таких, как алюминий и магний. Существенное значение имеет электролиз раствора NaCl с получением хлора, водорода и щелочи, а также электролитический способ производства ряда препаратов (KMnO4, NaClO, бензидин, органические фторпроизводные и др.). Катодное осаждение металлов играет большую роль в металлургии цветных металлов и в технологии гальванотехники.
Поляризационные явления в химических источниках тока.
Для ОВР RedL + OxR → OхL + RedR в гальваническом элементе под током I , в соответствии с вольт-амперной характеристикой электродных полуреакций (рис. 5.11) анодная поляризация (перенапряжение) увеличивает равновесный потенциал, а катодная - уменьшает. (Ниже представлены модули перенапряжений.)
OxL + ze- ← RedL, φa = φp,a + ηa ,
OxR + ze- → RedR , φk = φp,k – ηk.
ЭДС гальванического элемента EI = φk – φa = φp,k – φp,a – (ηk + ηa ).
По закону Ома I = EI /( R + Rex )= [E – (ηk + ηa )] /( R + Rex ),
R, Rex - сопротивление самого источника и внешней нагрузки.
Сопоставление поляризационных кривых 1, 2 и 1', 2' на рис. 5.12 (II) показывает, что чем выше поляризация, тем меньше отбираемый ток и мощность.
Снижение вредных поляризационных явлений при разработке гальванического элемента осуществляется соответствующим выбором систем и введением специальных деполяризаторов.