

nanotechnology / apl76(..)00
.pdfAPPLIED PHYSICS LETTERS |
VOLUME 76, NUMBER 25 |
19 JUNE 2000 |
Electric-®eld dependence of mobility in conjugated polymer ®lms
S. V. Rakhmanova and E. M. Conwella)
Center for Photoinduced Charge Transfer, Chemistry Department, University of Rochester,
Rochester, New York 14627
~Received 4 January 2000; accepted for publication 20 April 2000!
It has been suggested that Poole±Frenkel behavior, i.e., the linear dependence of the log of the mobility m on the square root of the electric-®eld intensity, found for many conjugated polymer ®lms is due to the interaction between the charge carriers and randomly distributed permanent dipoles. However, the dipole concentration in the polymers is much too small to achieve this. We show that introduction of short-range correlation between the energies of the hopping sites, on the scale of the coherence length measured by x rays, can account for the ®eld and temperature dependence of ®lms of poly~2-methoxy,5-~28-ethyl-hexoxy!±p-phenylene vinylene!, a representative conjugated polymer. We investigate also the effects of inhomogeneity on the ®eld and temperature dependence of m. © 2000 American Institute of Physics. @S0003-6951~00!02625-5#
Conjugated polymers have become important because of their clear possibilities for display applications. A great deal of work has been done on polymer light-emitting diodes ~PLEDs! for such applications. Among the properties of the PLEDs still in need of improvement are electron and hole transport. Given a set of Ohmic or near-Ohmic contacts, which it is known, at least in principle, how to achieve, the current±voltage (I ± V) characteristic, and in turn the light output, of the PLED are determined by the carrier transport. Mobility values for holes and electrons in ®lms of these materials have been obtained from measurements of time of ¯ight and by deduction from space-charge-limited currents. It has been found that mobility m of electrons and holes in ®lms of p-phenylene vinylene and many of its derivatives varies with ®eld E over a wide range of ®elds according to the Poole±Frenkel ~PF! law, i.e.,
m5m0 exp~gAE 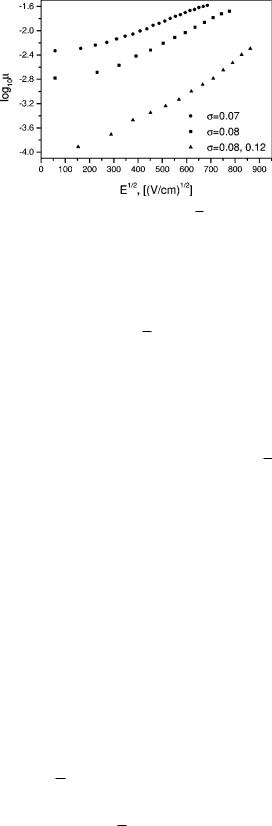
Appl. Phys. Lett., Vol. 76, No. 25, 19 June 2000 |
S. V. Rakhmanova and E. M. Conwell |
3823 |
ergy of spatially close sites should be correlated. Crystalline regions or aggregates are examples of regions that should have smaller spacing and correspondingly larger polarization energy, leading to lower site energies; amorphous or disordered regions with presumably larger spacing should have smaller polarization energy and thus higher site energies. As mentioned above, another source of variation of site energies in polymers is that, outside the long conjugation length limit, the energy of an electron or hole added to ~hopping onto! a conjugation length depends on the length. More-ordered regions have generally been found to have longer conjugation lengths. Thus, it appears likely that regions with a similar degree of order or disorder, or under compression or expansion, should have similar, i.e., to some extent correlated, site energies.
We will show now, through the use of Monte Carlo simulations including correlation, that it is possible to account for the experimental observation of PF behavior in MEH±PPV. The simulations were performed for a cubic lattice of 20032003200 sites with spacing a between nearest neighbors. Periodic boundary conditions are applied in all three directions. Most of the calculations were done for a 53 nm, which is of the order of the average conjugation length and also the value found for the average hopping dis-
tance in PPV from ®tting the transit time of the fastest carriers to the Scher±Montroll scaling law.15,16 The site ener-
gies were taken from a Gaussian distribution. The probability of a site having energy « is given by (2ps2)21/2 exp
(2«2/s2), where s is the variance.
We studied two types of samples, ones with s constant throughout ~homogeneous!, others with two values of s ~inhomogeneous!. The inhomogeneous samples had 20% of the regions with variance s1 set either regularly or randomly, in a matrix of variance s0 . After a sample was prepared by distributing the site energies «k with appropriate s, we introduced short-range correlation between the site energies as in Ref. 6, taking the energy of the ith site as the average of the energies of all the sites contained within a sphere of radius a, i.e., «i5N (k5nn«k , where N is a normalization factor and nn stands for nearest neighbors. Note that a correlation length of a53 nm is a little less than the coherence length found by x-ray diffraction for MEH±PPV ~Ref. 13! and earlier for PPV.17
The jump rate ni j was taken as n0 exp(2D/kT), where n0 is a scale factor and D, following the Miller±Abrahams formulation, is the energy difference between sites, including the contribution of the ®eld, for uphill jumps, and 0 for downhill jumps.18 The dwelling time on site i is determined from ti5(S j ni j )21. The destination of an actual hop is determined by a random number generator in accordance with the relative magnitudes of the ni j . Each particle was allowed to make up to 23105 hops. If a particle has advanced more than 1500 sites in the direction of the ®eld its motion is terminated, provided it has made at least 53104 hops. Knowing the total time of travel and the distance the particle traveled along the ®eld direction, we can determine its average velocity. To obtain the drift velocity we averaged over 40 particles started from randomly chosen sites, and over a number of realizations of the disorder. We found that usually four realizations were enough for small variances (s
FIG. 1. 290 K Monte Carlo results for log10 m vs AE for two homogeneous samples ~circles for s50.07; squares for s50.08! and one inhomogeneous sample ~triangles! with the values of s indicated, and correlation described by Eq. ~1!.
,0.1 eV), but for larger variances averaging over more samples ~up to 14 in some cases! was required to accumulate better statistics.
Some results for log m vs AE at 290 K are shown in Fig. 1 for homogeneous samples with different values of s. It is seen that larger s is correlated with smaller mobility, as expected because larger s implies the presence of some larger barriers. The change of mobility when s goes from 0.07 to 0.08 eV is a factor 2 at E533105 V/cm. With increase in s, g increases, the effect of the ®eld being larger when there are larger barriers. However, the increase in g with increasing s is gradual. The departure from linearity of log m vs AE seen at high ®elds can be attributed to the decreasing effect of the barriers. When the ®eld is high enough a carrier can pick up in one hop energy comparable to s, and thus to the average barrier height, making the barriers less effective.
Also shown in Fig. 1 is a plot for a sample with inhomogeneous disorder, 80% of the sample having s 50.08 eV, the other 20% having s50.12 eV distributed either randomly or in a single layer. Signi®cantly, despite the inhomogeneity, the PF behavior is maintained over a range comparable to that for the homogeneous samples, and g is not much increased. The mobility, however, is greatly decreased. Thus, increasing s by 50% in 20% of the sample, however distributed, decreased m by more than a factor 10.
The results of the Monte Carlo simulations are in good overall agreement with experiment. For s50.07 eV, log m
varies linearly |
with A |
|
over the range 73104,E,4 |
E |
|||
3105 V/cm. |
In that |
®eld range we ®nd g53.5 |
|
31023~cm/V!1/2. These ®gures agree well with the time-of- ¯ight data for holes in MEH±PPV of Campbell et al.,19 who found log m}AE in the range 53104 ± 53105 V/cm with
E058.73104 V/cm, |
corresponding |
to |
g53.4 |
31023(cm/V)1/2. For |
s50.08 eV m is |
smaller, |
the ®eld |
range over which log m}AE is shifted up a little, to 13105
up to 63105 V/cm, and g is a little larger, speci®cally, 4.6 31023(cm/V)1/2. The ®eld range and g for s50.08 eV are
in good agreement with the ®ndings for holes in MEH±PPV obtained by measurement of space-charge-limited current, g being 4.831023(cm/V)1/2.8 It must be noted, however, that the apparently good agreement between the experimental results and the simulations for homogeneous samples does not preclude the samples being inhomogeneous because, as dis-
Downloaded 27 Nov 2006 to 146.103.254.11. Redistribution subject to AIP license or copyright, see http://apl.aip.org/apl/copyright.jsp
3824 |
Appl. Phys. Lett., Vol. 76, No. 25, 19 June 2000 |
S. V. Rakhmanova and E. M. Conwell |
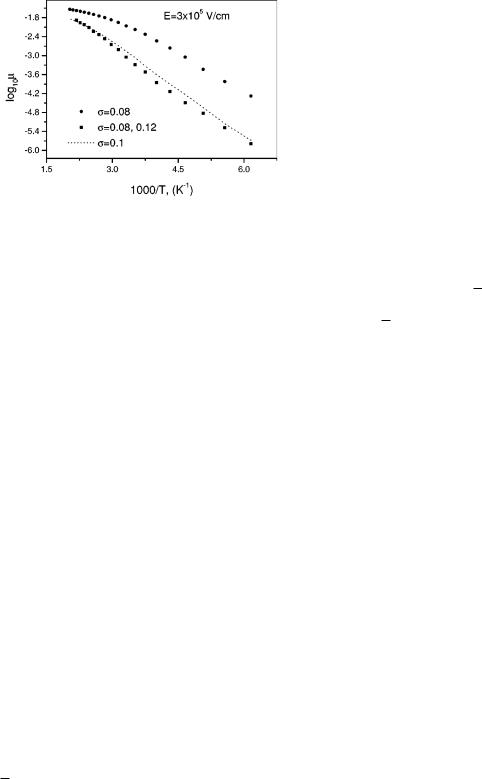
FIG. 2. 290 K Monte Carlo results for log10 m vs 1000/T for two homogeneous samples ~circles for s50.08; dashed line for s50.1! and one inhomogeneous sample ~triangles! for a ®eld of 23105 V/cm and correlation described by Eq. ~1!.
cussed in the last paragraph, inhomogeneity may not greatly change g and the ®eld range of PF behavior. We will show below that there is some evidence for inhomogeneity in these samples.
The temperature dependence of log m obtained by Monte Carlo simulation for some of the cases we have been discussing is shown in Fig. 2. As is seen in Fig. 2, in the temperature range where measurements are usually made, ;200± 320 K, the log m data for the homogeneous samples do not fall on a straight line when plotted versus 1000/T. They do, however, fall on a straight line when plotted versus (1000/T)2, as predicted by the disorder theory.5 In the data for the inhomogeneous sample the situation is reversed; log m falls on a straight line when plotted versus 1000/T, i.e., shows activated behavior, as seen in Fig. 2, but not when plotted versus (1000/T)2. There are many instances in the conjugated polymer literature where activated behavior has been found, for example, in Refs. 8 and 20 for PPV derivatives and in Ref. 21 for a ladder polymer. We suggest that the activated behavior is indicative of sample inhomogeneity.
It is of interest to compare the slope of log m vs 1000/T obtained from our Monte Carlo calculations for the inhomogeneous sample with that obtained experimentally for MEH± PPV. The temperature dependence of m arises from both m0 and g. m0 has been found to increase exponentially with T according to exp(2D/kT), with D50.3860.02 eV for holes in MEH±PPV.11 Experimentally, the plot of g vs 1/kT has
been found to be a straight line with slope, denoted B, (2.3 60.2)31024 eV~cm/V!1/2 for holes in MEH±PPV.11 Com-
bination of these results gives the slope of log m vs 1/kT as (BAE2D), which, for E533105 V/cm, is 20.25 eV for MEH±PPV with the numbers just given. For the inhomogeneous sample of Fig. 2 the slope of log m vs 1/kT is 20.21 eV, quite comparable. Exact agreement could not be expected in any case because g is not the same for the two samples.
Finally, we consider how the results we have derived can explain the apparently anomalous behavior of a ladder polymer sample MeLPP. The weak temperature and ®eld dependence of the time-of-¯ight mobility of this sample, designated sample 1 in Ref. 21, indicate a small value of s, perhaps 0.04 or less. The fact that the mobility is much smaller than expected for such a small s could be explained by the existence of large s in some portion of the sample which, as shown by the results in Fig. 1 can decrease the mobility drastically. Additional evidence for inhomogeneity of this sample is the dependence, shown in Fig. 7 of Ref. 21, of log m on 1/T rather than 1/T2.
In summary, we have shown that correlation, on the scale of the x-ray measured coherence length, between the energies of the hopping sites can account for the observed Poole±Frenkel dependence of mobility on ®eld in MEH± PPV, and, by inference, other conjugated polymers. Inhomogeneity, embodied by spatial variation in s in our calculations, still allows log m}AE, but can account for variations between samples in the size of the mobility and the coef®- cient of AE. It can also account for the observation for many samples that log m}1/T rather than the predicted 1/T2.
The support of the National Science Foundation under Science and Technology Center Grant No. CHE912001 is gratefully acknowledged.
1P. M. Borsenberger and J. Shi, Phys. Status Solidi B 191, 461 ~1995!.
2P. M. Borsenberger, W. T. Gruenbaum, and E. H. Magnin, Physica B 228, 226 ~1996!.
3L. B. Schein, A. Peled, and D. Glatz, J. Appl. Phys. 66, 686 ~1989!.
4W. G. Gill, J. Appl. Phys. 43, 5033 ~1972!.
5For a review, see H. Bassler, Phys. Status Solidi B 175, 15 ~1993!.
6Yu. N. Gartstein and E. M. Conwell, Chem. Phys. Lett. 245, 351 ~1995!.
7S. V. Novikov, D. H. Dunlap, V. M. Kenkre, P. E. Parris, and A. V. Vannikov, Phys. Rev. Lett. 81, 4472 ~1998!.
8L. Bozano, S. A. Carter, J. C. Scott, G. G. Malliaras, and P. J. Brock, Appl. Phys. Lett. 74, 1132 ~1999!.
9G. G. Malliaras, J. R. Salem, P. J. Brock, and J. C. Scott, Phys. Rev. B 58, R13411 ~1998!.
10S. V. Novikov, D. H. Dunlap, V. M. Kenkre, P. E. Parris, and A. V. Vannikov, Phys. Rev. Lett. 81, 4472 ~1998!.
11J. Cornil, D. Beljonne, and J.-L. Bredas, J. Chem. Phys. 103, 842 ~1995!.
12H. A. Mizes and E. M. Conwell, Synth. Met. 68, 145 ~1995!.
13C. Y. Yang, F. Hide, M. A. Diaz-Garcia, A. J. Heeger, and Y. Cao, Polymer 39, 2299 ~1998!.
14T-Q. Nguyen, I. Martini, J. Liu, and B. J. Schwartz, J. Phys. Chem. B 104, 237 ~2000!.
15P. W. M. Blom and M. C. J. M. Vissenberg, Phys. Rev. Lett. 80, 3819 ~1998!.
16H. C. F. Martens, H. B. Brom, and P. W. M. Blom, Phys. Rev. B 60, R8489 ~1999!.
17D. A. Halliday, P. L. Burn, D. D. C. Bradley, R. H. Friend, O. M. Gelsen, A. B. Holmes, A. Kraft, J. H. F. Martens, and K. Pichler, Adv. Mater. 5, 40 ~1993!.
18A. Miller and E. Abrahams, Phys. Rev. 120, 745 ~1960!.
19I. H. Campbell, D. L. Smith, C. J. Neef, and J. P. Ferraris, Appl. Phys. Lett. 74, 2809 ~1999!.
20P. W. M. Blom, M. J. M. de Jong, and M. G. van Munster, Phys. Rev. B 55, R656 ~1997!.
21D. Hertel, H. Bassler, U. Scherf, and H. H. Horhold, J. Chem. Phys. 110, 9214 ~1999!.
Downloaded 27 Nov 2006 to 146.103.254.11. Redistribution subject to AIP license or copyright, see http://apl.aip.org/apl/copyright.jsp