

5 курс / Госпитальная педиатрия / Атлас_редких_болезней_Педиатрия,_2016_г_
.pdfАтлас редких болезней
В |
Г |
В. На компьютерной томографии высокого разрешения больной муковисцидозом, 7 лет, на фоне значительной деформации легочного рисунка с наличием распространенных бронхо-
и бронхиолоэктазов в S3 верхней доли правого легкого. Вдоль междолевой плевры нижнего язычкового сегмента (S5) слева определяются зоны фиброзных изменений легочной ткани
Г. Компьютерная томография высокого разрешения больной муковисцидозом, 12 лет, вне обострения процесса. Распространенные, но умеренные бронхоэктатические изменения с малой степенью выраженности перибронхиальной инфильтрации. Практически отсутствуют мукоидные пробки.
В эмфизематозные изменения вовлечены не более 5 сегментов с каждой стороны
Базисное лечение. Больной муковисцидозом должен получать лечение
втечение всей жизни. Большинство пациентов, особенно при хронической синегнойной инфекции и тяжелом течении заболевания, нуждаются в курсах внутривенной антибактериальной терапии каждые 3–4 месяца: вводятся препараты в зависимости от чувствительности микрофлоры в течение 14–28 дней. Проникновению антибиотиков в колонии псевдомонад способствует применение азитромицина: его вводят альтернирующими длительными курсами, 3 раза
внеделю по 1 дозе (10 мг/кг).
Ингаляционные антибиотики. Для базисной терапии при хроническом высеве синегнойной палочки используют специальные растворы антибиотиков (тобрамицин, полимиксин) для небулайзерной терапии. Инновационной формой в терапии синегнойной инфекции является порошковая форма тобрамицина (Тоби Подхалер).
Противовоспалительная терапия. Подавить полностью воспалительный процесс в легких с помощью антибиотиков удается не всегда. С этой целью проводят альтернирующие курсы ибупрофена и других нестероидных противовоспалительных препаратов, а также глюкокортикостероидов (редко, по особым показаниям).
Муколитическая терапия является обязательной для всех больных муковисцидозом. Применяются препараты N-ацетилцистеина, амброксола гидрохлорида
и карбоцистеина: внутрь и в виде ингаляций. Муколитики из различных групп применяются в сочетании, например ацетилцистеин внутрь + ингаляции амброксола.
160
Муковисцидоз
Ж
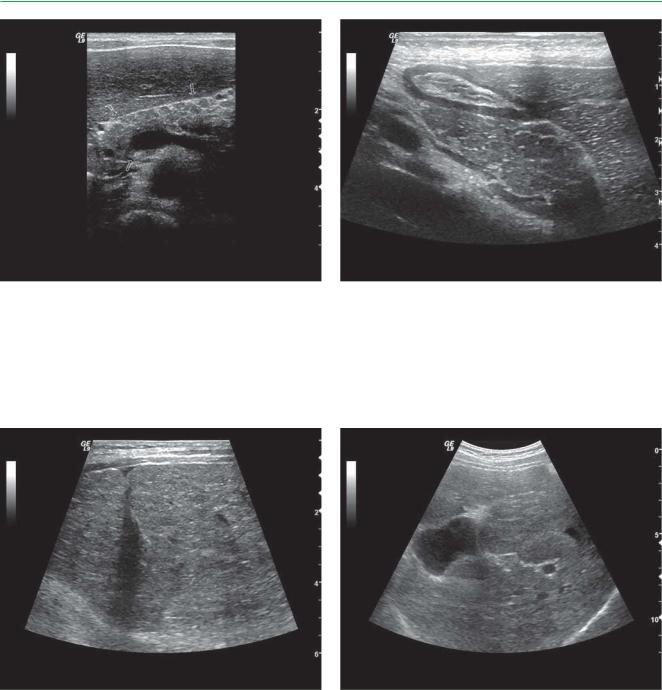
Эхограмма поджелудочной железы |
Эхограмма поджелудочной железы (обозначена |
(обозначена стрелками) ребенка 4 лет |
стрелками) ребенка 8 лет с муковисцидозом, |
с муковисцидозом, поперечный срез. |
продольный срез. В паренхиме поджелудочной |
В паренхиме поджелудочной железы определяются |
железы определяются множественные кисты — |
множественные кисты — гипоэхогенные |
гипоэхогенные образования овальной формы |
образования овальной формы с четким контуром |
с четким контуром. «Ж» — желудок с утолщенными |
|
стенками |
ЖП
Эхограмма печени (косой срез) ребенка 13 лет |
Эхограмма печени (косой срез) ребенка 14 лет |
с муковисцидозом. Контур печени неровный, |
с муковисцидозом. Эхогенность паренхимы |
эхогенность паренхимы повышена, выраженная |
повышена, выраженная неоднородность |
неоднородность паренхимы из-за множественных |
паренхимы из-за множественных крупных |
крупных и средних участков повышенной |
участков повышенной эхогенности. «ЖП» — |
эхогенности |
деформированный, увеличенный желчный пузырь |
Наиболее эффективным муколитиком при муковисцидозе является рекомбинантная человеческая дезоксирибонуклеаза — дорназа альфа (Пульмозим): постоянно с момента постановки диагноза.
Используют гипертонический раствор: 5–7% NaCl по 4 мл 2–3 раза в день через небулайзер.
161
Атлас редких болезней
Бронхолитики. Бронхоспазм, особенно при легочной форме заболевания, встречается практически у всех больных муковисцидозом. С бронхолитической целью обычно применяются комбинированные препара-
ты |
(например, раствор фенотерола |
|
и ипратропия бромида) в виде небу- |
||
лайзерных ингаляций 2–3 раза в день. |
||
С целью профилактики постнагрузоч- |
||
ного бронхоспазма ингаляцию с брон- |
||
холитиком проводят непосредственно |
||
перед занятиями кинезитерапией. |
||
Эхограмма печени (косой срез) ребенка 10 лет |
Специальная кинезитерапия |
|
с муковисцидозом. Эхогенность паренхимы |
||
при муковисцидозе |
||
умеренно повышена, отмечается мелкоочаговая |
||
неоднородность |
Кинезитерапия — комплекс специ- |
|
альной дыхательной гимнастики и общей лечебной физической культуры, который показан всем детям с муковисцидозом.
Ферменто- и диетотерапия. Заместительная терапия нарушенной функции поджелудочной железы проводится панкреатическими ферментами в виде минимикросфер при каждом приеме пищи (в среднем 5000 единиц Ед липазы/кг веса в сут). Рацион по калорийности должен превышать на 20–50% возрастную норму. Следует подсаливать пищу (1–5 г в сут). Необходимо обеспечить прием достаточного количества жидкости, особенно детям грудного возраста.
Достижение и поддержание нормального нутритивного статуса у пациентов с МВ зачастую затруднено, в то же время это тесно коррелирует с прогнозом и выживанием. Широкий ассортимент смесей и дополнительные методы
сиспользованием ферментов поджелудочной железы могут быть эффективны при МВ.
Полуэлементная формула Peptamen (Nestle) без дополнительного приема ферментов усваивается также хорошо, как стандартная (полимерная) формула Isocal
сдополнительной заместительной терапией ферментами у больных муковисцидозом с недостаточностью функции поджелудочной железы. Продукты для для детей с МВ были специально разработаны Peptamen Junior и Peptamen Junior Advance. В состав смесей входит гидролизованный белок молочной сыворотки и СЦТ для лучшего усвоения и эффективной абсорбции.
Лечение обострений. Основа лечения обострений муковисцидоза — внутривенная антибактериальная терапия: курсы не меньше 3–4 недель, должны состоять из двух препаратов в максимальных дозах (например, два препарата внутривенно и один в ингаляциях).
Витаминотерапия. Всем больным необходимо постоянное дополнительное введение жирорастворимых витаминов: А, D, Е и К. Детям с дефицитом веса дополнительно вводится лечебное питание в виде высококалорийных смесей.
162

Муковисцидоз
Урсодезоксихолевая кислота применяется постоянно для профилактики холестаза, цирроза печени.
Новые виды терапии. Активно разрабатываются методы воздействия на клетку при разных классах дефекта белка МВТР (персонифицированная фармакогенетическая терапия), определенные успехи получены в ходе многоцентровых рандомизированных исследований, в частности одобрен к применению ивакафтор, показанный для пациентов с мутацией G551D (с 2014 года дополнительно еще и при мутациях G178R, S549N, S549R, G551S, G1244E, S1251N, S1255P
иG1349D).
Вфеврале 2016 года планируется расширить этот спектр за счет следующих мутаций-: 2789+5G- > A, 3849+10kbC->T, 3272–26A->G, 711+3A->G, E56K, P67L, R74W, D110E, D110H, R117C, L206W, R347H, R352Q, A455E, D579G, E831X, S945L, S977F, F1052V, R1070W, F1074L, D1152H и D1270N.
Второй молекулой, одобренной к применению у пациентов с МВ (генотип F508/F508) является сочетание Люмакафтора и Ивакафтора.
Диспансерное наблюдение. Диспансерное наблюдение за больными муковисцидозом должно проводиться при участии специалистов специализированных центров муковисцидоза.
Прогноз. Прогноз муковисцидоза остается серьезным, однако, чем раньше ставится диагноз и начинается базисная терапия, тем прогноз благоприятней. В последние годы с увеличением продолжительности жизни больные муковисцидозом занимают определенную социальную нишу и ведут активный образ жизни (учатся в школе, вузе, работают, создают семьи).
Клинический пример
Девочка А., 31.12.2000 года рождения (см. рис. на стр. 152).
Из анамнеза: родилась от 3-й беременности (1-я — в 1990 г., ребенок с диагнозом гемморагического колита умер в возрасте 4 мес; 2-я — девочка, 17 лет, здорова), протекавшей без особенностей, 3-х срочных родов. Масса тела при рождении 3400 г, длина тела 52 см. Оценка по шкале APGAR 8 баллов. Приложена к груди на 1-е сут, естественное вскармливание — до 14 дней. БЦЖ — в роддоме, ревакцинация не проводилась. Реакция Манту: в 3 года папула 10 мм, в 4 года — 11 мм, с 5 до 8 лет — не проводились, в 9 лет — отрицательная. Прививки по возрасту до 4 лет, далее не проводились. Аллергия на шерсть кошки (отек Квинке)
имандарины.
С2,5 мес отмечается жидкий частый стул. Наблюдалась гастроэнтерологом по поводу «Дисбактериоза». В возрасте 8 мес в связи с рвотой и жидким стулом госпитализирована в инфекционную больницу по месту жительства. В течение первого года жизни отмечено 2 эпизода пневмонии, в дальнейшем — частые бронхиты, пневмонии 1 раз в год (антибактериальная, симптоматическая терапия). Массу тела набирала плохо, постоянно наблюдается амбулаторно гастроэнтерологом.
В возрасте 4,5 лет, по данным копрологии, много нейтрального жира. При обследовании в стационаре по месту жительства была исключена целиакия, про-
163

Атлас редких болезней
ведены трижды потовые пробы — положительно (от 76 до 86 ммоль/л). Однако, диагноз «Муковисцидоз» выставлен не был.
С 5,5 лет болеет реже.
Ухудшение состояния с 7 лет: появились кашель, утомляемость, слабость, тошнота, перестала прибавлять в весе. Принимала фтивазид в течение 3 нед, усилилась тошнота. По данным рентгенографии была госпитализирована: остаточные явления пневмонии (назначен внутривенно цефтриаксон). Через 3 нед — повторное ухудшение состояния, кашель (повторная антибиотикотерапия, инъекции преднизолона — с временным улучшением). Выявлено значительное увеличение печени; в 4 раза превышена норма трансаминаз. Через полгода — два обострения с усилением кашля, подъемом температуры до фебрильных цифр, появилась потливость. Обследована в противотуберкулезном диспансере по месту жительства, при посеве мокроты выявлены кислотоустойчивые микобактерии. Проведена терапия цефотаксимом, гентамицином, амикацином, изониазидом (10% 2 мл в/м) под контролем ГУ ЦНИИТ г. Москвы. Туберкулез был исключен, посев на МБТ — отрицательный. Далее обследование продолжено в НЦЗД. На КТ органов грудной клетки выявленные изменения (бронхоэктазы, признаки обструктивного бронхита, участки пневмофиброза). Проведен потовый тест на аппарате «Макродакт»: электролиты пота 126 ммоль/л — результат положительный. Диагноз муковисцидоза впервые был выставлен в возрасте 9 лет. Подтвержден генетически: гомозигота по мутации delF508. В мокроте рост Pseudomonas aeruginosa, много нейтрального жира в кале, панкреатическая эластаза-1 кала: 69 мкг/г (тяжелая степень панкреатической недостаточности).
Дополнительно: на основании данных анамнеза, результатов лабораторноинструментальных обследований (положительный анализ крови на антитела к глиадину и тканевой трансглютаминазе, результаты биопсии тощей кишки) был выставлен сопутствующий диагноз: «Целиакия».
Объем базисной терапии
•Безглютеновая диета + дополнительное специальное питание;
•Креон 10000: по 8 капсул в день, Креон 25000: по 9 капсул в день (в зависимости от вида и объема пищи);
•Пульмозим: 1 ингаляция в день через небулайзер;
•Урсосан: по 3 капсулы на ночь (750 мг);
•Сумамед: 125 мг по схеме 3 раза в нед;
•Лазолван: 1 табл. 2 раза в день;
•витамин Е: по 200 МЕ в день;
•витамин D2: 1 капля в день;
•Викасол: 1 табл. в нед;
•Капотен: 0,25 по 1/2 табл. 3 раза в день (умеренная легочная гипертензия по данным эхографии сердца);
•ингаляции колистина: по 1 млн ЕД 2 раза в день в течение 8 мес в постоянном режиме; затем ингаляции с раствором тобрамицина: 300 мг 2 раза в день (28 дней, 28 дней перерыв, несколько курсов).
За время наблюдения в течение 5 лет девочка хорошо прибавила в весе — 25 кг (в начале наблюдения в возрасте 9 лет вес составлял 19 кг, в 14 лет — 46 кг), значи-
164

Муковисцидоз
тельно улучшились показатели флоуметрии (ОФВ1 в 9 лет — 70%, 14 лет — 99%). В настоящее время девочка продолжает наблюдаться в Центре, соблюдает все рекомендации, хорошо развивается, активно занимается спортом, учится в школе.
Список рекомендованной литературы
1.Муковисцидоз. Современные достижения и актуальные проблемы. Под ред. Н.И. Капранова, Н.Ю. Каширской. Методические рекомендации. 4-е издание, переработанное и дополненное. Москва. 2011. 124 с.
2.Хрущев С.В., Симонова О.И. Физическая культура детей с заболеваниями органов дыхания. Учебное пособие для студентов высших учебных заведений.
Москва: Издательский центр «Академия». 2006. 304 с.
3.Doering G., Hoiby N. Ранняя терапия и профилактика поражения легких. Европейский консенсус. J of Cystic Fibrosis. 2004; 3 (2): 67–91.
4.Стандарты терапии больных муковисцидозом: Европейский консенсус. J of Cystic Fibrosis. 2005; 4: 7–26.
5.Hodson M., Geddes D., Bush A. Cystic Fibrosis. Third ed. by London: Edward Arnold (Publishers) Ltd. 2007. 477 р.
165
МУКОПОЛИСАХАРИДОЗЫ
(MUCOPOLYSACCHARIDOSIS)
МКБ-10: E76
Общие сведения
Мукополисахаридозы относятся к лизосомным болезням накопления наряду со сфинголипидозами, ганглиозидозами, гликопротеинозами, а также болезнями лизосомного транспорта и нейрональными цероид-липофусцинозами. Обусловлены данные заболевания мутациями генов, контролирующих процесс внутрилизосомного гидролиза макромолекул.
Первые сведения о мукополисахаридозах относятся к 1917 г., когда канадский доктор Чарльз Хантер описал у двух братьев в возрасте 8 и 10 лет развившиеся изменения опорно-двигательного аппарата, гепатосплено- и кардиомегалию, тугоухость, а также незначительное снижение интеллекта. Два года спустя немецкий педиатр Гертруда Гурлер наблюдала похожую, но более тяжелую картину заболевания у двух мальчиков, не состоявших в кровном родстве.
Классификация
Согласно ферментативным дефектам и тяжести клинической симптоматики, выделяют следующие типы мукополисахаридозов (МПС):
МПС I
•МПС I H — синдром Гурлер
•МПС I H/S — синдром Гурлер–Шейе
•МПС I S — синдром Шейе
МПС II — синдром Хантера, умеренная и тяжелая формы МПС III — синдром Санфилиппо
•МПС IIIА типа
•МПС IIIВ типа
•МПС IIIС типа
•МПС IIID типа
•МПС IIIE типа
МПС IV — синдром Моркио
•МПС IVА — синдром Моркио А
•МПС IVВ — синдром Моркио В
МПС VI — синдром Марото–Лами, умеренная и тяжелая формы МПС VII — синдром Слая
МПС IX
Все типы наследуются по аутосомно-рецессивному типу за исключением синдрома Хантера (тип наследования рецессивный, сцепленный с полом — Х-хромосомой).
166

Мукополисахаридозы
МУКОПОЛИСАХАРИДОЗ ТИП I
(MUCOPOLYSACCHARIDOSIS TYPE I)
МКБ-10: E76.2
Определение. Мукополисахаридоз тип I — наследственная лизосомальная болезнь накопления, характеризующаяся гетерогенными клиническими проявлениями: задержкой роста, умственной отсталостью, сердечно-легочными нарушениями, гепато-спленомегалией, множественным дизостозом, помутнением роговицы. Все вышеперечисленные признаки приводят к инвалидизации и летальному исходу при тяжелом течении болезни.
Эпидемиология. МПС тип I встречается с популяционной частотой 1:40000– 100000.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Патология обусловлена гетерогенной группой мутаций в гене, кодирующем лизосомный фермент альфа-L-идуронидазу. Ген альфа-L- идуронидазы (IDUA) картирован на 4p16.3.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
4p16.2 |
|
|
|
|
4p15.31 |
|
|
|
4p13 |
|
|
|
4q13.3 |
|
4q21.23 |
|
|
4q22.3 |
|
|
|
|
4q26 |
|
|
|
|
4q28.3 |
|
|
4q31.23 |
|
|
|
4q32.3 |
|
|
|
|
4q34.3 |
|
|
||||||||||||||||
|
4p15.33 |
|
|
4p15.1 |
|
4q13.1 |
4q21.21 |
4q22.1 |
|
4q24 |
|
|
4q28.1 |
|
4q31.21 |
4q32.1 |
|
|
4q34.1 |
|
4q35.2 |
|||||||||||||||||||||||||||||||||||||
Ген альфа-L-идуронидазы
Дефицит альфа-L-идуронидазы может привести к развитию разных фенотипов болезни. В настоящее время выделяют три фенотипа болезни: синдром Гурлер (мукополисахаридоз I H — тяжелая форма), синдром Шейе (мукополисахаридоз I S — легкая форма) и синдром Гурлер–Шейе (мукополисахаридоз I H/S — промежуточная форма).
МУКОПОЛИСАХАРИДОЗ I H (ТЯЖЕЛАЯ ФОРМА)
(MUCOPOLYSACCHARIDOSIS TYPE H)
OMIM 607014
Синонимы: синдром Гурлер, Пфаундлера–Гурлер синдром.
Впервые описан в 1919 г. немецким детским врачом Г. Гурлер (G. Hurler).
167
Атлас редких болезней
Эпидемиология. МПС тип I встречается с популяционной частотой 1:20000– 100000 новорожденных.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. В основе болезни лежат мутации гена альфа-L- идуронидазы (IDUA) (картирован на 4р16.3), приводящие к отсутствию альфа-L- идуронидазы, которая является лизосомальной гидролазой — главным ферментом катаболизма мукополисахаридов. Это приводит к аккумуляции гепарансульфата и дерматансульфата. В эпителиальных и мезенхимных тканях накапливаются гликозаминогликаны (ГАГ), в нервных тканях — липиды.
Клинические проявления
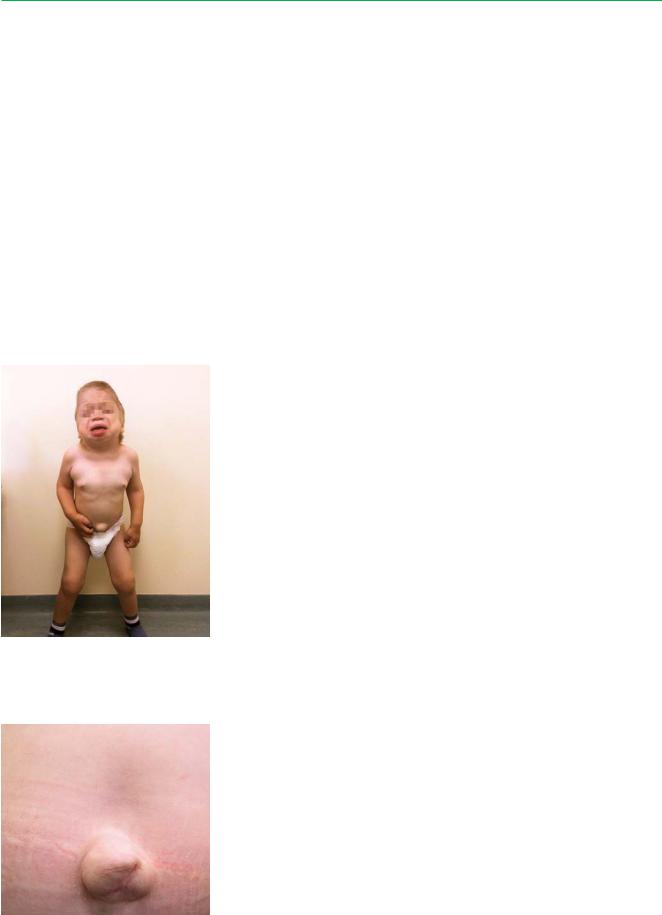
Внешний вид. Грубые черты лица, умственная отсталость, помутнение роговицы, тугоподвижность в суставах.
После рождения ребенок выглядит нормальным, симптоматика чаще всего развивается послe 1 года в виде задержки роста, которая начинается с 6–18 месяцев. У детей низкий рост с пропорциональным телосложением, скафоцефалия, макроцефалия, грубые черты лица, пухлые губы, широкие ноздри, запавшая переносица, орбитальный гипертелоризм, маленькие редкие зубы, макроглоссия, короткая шея.
Также встречаются гипертрихоз, гепатоспленомегалия, нарушение слуха, пупочная грыжа, паховая грыжа.
На поздних стадиях развития болезни — глухота, слепота, глубокая деменция.
|
Костная система. Максимальный рост 110 см. |
|
|
Обращает на себя внимание широкая грудная клетка, |
|
|
уменьшение подвижности в крупных и мелких суста- |
|
Пациент К., 6 лет 10 мес. |
вах, грудопоясничный кифоз с образованием горба. |
|
Рентгенография грудной клетки: укорочение |
||
Низкий рост (102 см). Синдром |
||
апноэ тяжелой степени |
и расширение диафизов трубчатых костей. Упло- |
|
|
щение и расширение турецкого седла, клювовид- |
|
|
ная форма тел позвонков. Ортопедические ослож- |
|
|
нения приводят к выраженному болевому синдрому |
|
|
и неподвижности. |
|
|
Органы дыхания: частые респираторные заболе- |
|
|
вания в виде ринитов, отитов. |
|
|
Накопление ГАГ в миндалинах, надгортаннике, |
|
|
трахее приводит к утолщению и сужению дыхатель- |
|
|
ных путей, развитию обструктивного апноэ. |
|
|
Орган зрения: помутнение роговицы, пигментная |
|
|
дегенерация роговицы, глаукома. |
|
Пациент К., 6 лет 10 мес. |
Центральная нервная система: задержка темпов |
|
Пупочная грыжа |
психоречевого развития, позже появляется глубокая |
168

Мукополисахаридозы
деменция; нарушение речи; туннельный синдром — синдром запястного канала, сообщающаяся гидроцефалия; пахименингит в шейном отделе, приводящий к сдавлению спинного мозга и последующей миелопатии.
Сердечно-сосудистая система характеризуется утолщением клапанов, сужением артерий, нарастающей ригидностью миокарда, кардиомиопатией; встречается артериальная гипертония. С возрастом может развиться сердечная недостаточность.
Желудочно-кишечная система: пахово-мошоночные и пупочные грыжи, гепатоспленомегалия.
Диагностика
•Характерный внешний вид пациента.
•Повышенная экскреция дерматансульфата и гепарансульфата с мочой, метахроматические гранулы в 10–60% лейкоцитов, метахроматическая окраска фибробластов.
•Отсутствие фермента с использованием искусственных субстратов в культуре фибробластов или изолированных лейкоцитов.
•Для пренатальной диагностики применяется метод измерения активности альфа- L-идуронидазы в культуре амниоцитов или в биоптате ворсинок хориона.
Дифференциальный диагноз:
•другие виды мукополисахаридозов;
•ганглиозидозы.
Прогноз. Смерть наступает на втором десятилетии жизни. В среднем продолжительность жизни пациентов составляет 11,6 лет. Наиболее частыми причинами смерти являются обструкции верхних дыхательных путей, сердечная недостаточность и инфекции дыхательных путей.
МУКОПОЛИСАХАРИДОЗ ТИП I S (ЛЕГКАЯ ФОРМА)
(MUCOPOLYSACCHARIDOSIS TYPE S)
OMIM 607015
Синонимы: синдром Шейе, клеточная метахромазия, Шпета–Гурлер синдром, синдром Ульриха–Шейе, дизостоз Моркио с помутнением роговицы, наследственная остеоартропатия с рецессивным наследованием, поздняя форма болезни Пфаундлера–Гурлер, Шинца (Schinz) синдром.
Впервые описана американским офтальмологом Гарольдом Шейе (Н.G. Scheic) в 1962 г.
Эпидемиология. Средняя частота 1:100000.
Тип наследования: аутосомно-рецессивный.
169