

5 курс / Госпитальная педиатрия / Атлас_редких_болезней_Педиатрия,_2016_г_
.pdfАтлас редких болезней
рующих клеточное деление, дифференцировку и миграцию, в том числе при посредничестве эпидермального фактора роста. Последний путь имеет весомое значение в формировании полулунных клапанов. Мутации этого гена также могут стать причиной синдрома Noonan, а также ряда лимфопролиферативных заболеваний.
Клинические проявления
Кожа — лентиго — пигментарные распространенные плоские или слегка возвышающиеся пятна на коже цвета кофе с молоком и/или темно-коричневые «кофейные» пятна диаметром 1,5–3 см, иногда сливающиеся, располагающиеся диссеминированно по всему кожному покрову, но, как правило, не затрагивающие лицо.
Сердечно-сосудистая система — стеноз легочной артерии и/или гипертрофическая кардиомиопатия (ГКМП), часто с поражением полулунных клапанов.
Орган зрения — широко расставленные глаза. Орган слуха — нейросенсорная глухота.
Репродуктивные органы — неопущение, дисплазия яичек, запоздалое появление mensis.
Нервно-психическое и физическое развитие — задержка роста и развития.
Изменения ЭКГ — одноили двухжелудочковая гипертрофия, нарушения проводимости, АВ-и внутрижелудочковые блокады.
Диагностика
Клиническая симптоматика. Диагностическими критериями синдрома LEOPARD (по Voron et al., 1976) являются:
•множественное лентиго + 2 других признака или
•наличие множественного лентиго у родственника 1-й линии родства + 3 других признака заболевания у пациента.
Cреди 8 пациентов с синдромом LEOPARD и установленными мутациями гена
PTPN11 лицевой дисморфизм отмечался в 100% случаев, ГКМП — в 87%, пятна «кофе с молоком» — в 75%.
Характерный фенотип: орбитальный гипертелоризм (100%), измененные ушные раковины (87%), низкопосаженные уши (50%). У 6 пациентов (75%) имели место аномалии грудной клетки.
Инструментальная диагностика Эхокардиография. По сравнению с пациентами с идиопатической ГКМП
у пациентов с ГКМП в структуре синдромов LEOPARD и Noonan эхокардиографические особенности заключаются в следующем: более выражена гипертрофия миокарда желудочков, в большей степени изменена диастолическая функция миокарда, чаще наблюдается обструкция выходного тракта левого желудочка, нередко в сочетании с обструкцией выходного тракта правого желудочка, дилатацией коронарных артерий и изменениями со стороны клапанного аппарата.
Подтверждение диагноза. Диагноз может быть подтвержден с помощью генетических тестов. Однако, по литературным данным, среди новорожденных пациентов, соответствующих клиническим критериям, при генетическом исследовании диагноз подтверждается только в 80% случаев.
250

Синдром LEOPARD
Дифференциальный диагноз:
•синдром Noonan;
•нейрофиброматоз 1-го типа.
Лечение. Специфического лечения не существует.
При значимом снижении уровня тиреотропного и фолликулостимулирующего гормонов рекомендована заместительная терапия.
При выраженных кожных проявлениях — криохирургическое лечение. Тяжелое поражение сердца требует профилактики жизнеугрожающих нарушений ритма и проводимости, при необходимости — хирургической коррекции.
Необходимо обследование всех членов семьи, в том числе и детей. При планировании беременности целесообразно генетическое консультирование с последующим проведением внутриутробной эхокардиографии.
Прогноз. Важнейшую роль в прогнозе у таких пациентов играет кардиальная патология. Описаны случаи позднего проявления поражения сердечнососудистой системы и развитие внезапной сердечной смерти у пациентов.
Список рекомендованной литературы
1.Bruce D. Gelb, Marco Tartaglia. Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Human Molecular Genetics. 2006; 15 (2): 220–226.
2.Digilio M. C., Conti E., Sarkozy A., Mingarelli R., Dottorini T., Marino B., Pizzuti A., Dallapiccola B. Grouping of Multiple-Lentigines. LEOPARD and Noonan Syndromes on the PTPN11 Gene. Am J Hum Genet. 2002; 71: 389–394.
3.Sarkozy A.,Conti E.,Digilio M.C.,Marino B.,Morini E.,Pacileo G.,Wilson M.,Calabro R., Pizzuti A., Dallapiccola B. Clinical and molecular analysis of 30 patients with multiple lentigines LEOPARD syndrome. J Med Genet. 2004; 41: 1–6.
4.Sarkozy A., Digilio M.C., Dallapicolla B. Leopard syndrome. Orphanet Journal of Rare Diseases. 2008; 3: 1–8.
5.Gibson W., Trevenen C., Giuffre M., Alexander K.C. Noonan syndrome in a premature infant with hypertrophic cardiomyopathy and death in infancy. J Natl Med Assoc. 2005; 97: 805–807.
6.Cerrato F., Pacileo G. et al. A standard echocardiographic and tissue Doppler study of morphological and functional findings in children with hypertrophic cardiomyopathy compared to those with left ventricular hypertrophy in the setting of Noonan and LEOPARD syndromes. Cardiol Young. 2008; 18: 575–580.
7.Madhusudan Ganigara, Atul Prabhu, Raghvannair Suresh Kumar. LEOPARD syndrome in an infant with severe hypertrophic cardiomyopathy and PTPN11 mutation. Ann Pediatr Cardiol. 2011; 4 (1): 74–76.
8.Martinez-Quintana E., Rodriguez-Gonzalez F. LEOPARD syndrome: clinical features and gene mutations. Mol Syndromol. 2012; 3 (4): 145–157.
9.Digilio M.C., Lepria F., Sopathies R.A. Clinical diagnosis in the first year of life. Mol Syndromol. 2011; 1 (6): 282–289.
251
СИНДРОМ АЛАЖИЛЛЯ
(ALAGILLE SYNDROME)
МКБ-10: Q44.7; ОМIМ 118450
Определение. Cиндром Алажилля — аутосомно-доминантное генетическое заболевание, поражающее печень, сердце, почки и другие системы организма.
Синонимы: артериопеченочная дисплазия, cиндромальная гипоплазия внутрипеченочных желчных путей, синдром Алажилля–Ватсона.
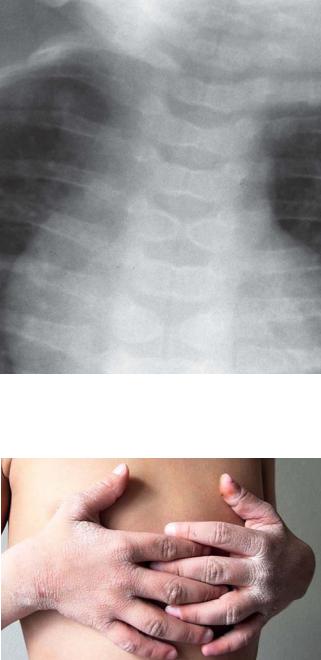
Рентгенограмма шейно-грудного отдела позвоночника больного М. 3 лет с синдромом Алажилля, дизостоз тел СIII–ТX
Больной К., 8 лет. Cиндром Алажилля нелеченый. Тотальный гиперкератоз кожи, зуд
Эпидемиология. Частота синдрома Алажилля составляет 1:30000 новорожденных, в США — 1:50000–100000.
Тип наследования: аутосомнодоминантный, примерно в 60% случаев выявляют новые мутации.
Этиология, патогенез. Болезнь обусловлена мутациями в гене 1 JAG1, картированном на коротком плече 20 хромосомы (20р12). Врожденный дефект эмбриогенеза с поражением нескольких систем.
Клинические проявления
Впервые описан Daniel Alagille в 1969 г. как заболевание с типичной комбинацией пяти основных признаков:
•холестазом, обусловленным дуктулярной билиарной недостаточностью;
•специфическим фенотипом — лицевым дисморфизмом;
•стенозом легочной артерии и/или ее ветвей и другими пороками сердца;
•незаращением тел преимуще-
ственно грудных позвонков с характерной картиной «бабочки» на рентгенограмме;
•офтальмологическими аномалиями — задним эмбриотоксоном (врожденный дефект роговицы).
252
Cиндром Алажилля реже сочетается с почечной дисплазией и/или кистами в почках, панкреатической недостаточностью и другими проявлениями.
Поражение печени — дуктулярная гипоплазия и хронический холестаз: характерный признак — зуд, вплоть до изнуряющего, гиперкератоз кожи гипербилирубинемия нетипична, в сыворотке крови повышено содержание желчных кислот, гамма-глютамил- транспептидаза может быть высокой и нормальной. Дефицит жирорастворимых витаминов и почечный канальцевый ацидоз приводят к вторичному синдрому Фанкони.
Поражение сердца: стеноз легочной артерии в большинстве случаев, 24% — другие пороки.
Лицевой дисморфизм: выступающий лоб, глубоко посаженные глаза, выступающий острый подбородок, седловидная переносица, нос прямой или «птичий».
Офтальмологические проявления: дефекты передней камеры глаза, задний эмбриотоксон, аномалии Аксенфельда–Ригера, ретинальная пигментопатия и эпителиопатия.
Аномалии скелета: незаращение тел позвонков. Поражение почек, задержка роста, панкреатическая
недостаточность, хронический средний отит, интракраниальные кровоизлияния.
Диагностика
Для фенотипической диагностики синдрома Алажилля достаточно 3 из 5 мажорных признаков.
Молекулярно-генетический анализ
Подавляющее большинство (94%) пациентов имеет мутации в гене Jagged1 (JAG1), расположенном на коротком плече 20-й хромосомы (20p12), и лишь неболь-
шая часть (< 1%) — мутации в NOTCH-лиганде того же гена, отвечающего за пролиферацию и дифференцировку разных клеток в процессе развития. Было установлено, что мутации Notch1 являются причиной поражения аортального клапана, Notch2 отвечают за нормальный эмбриогенез, мутации Notch3 становятся причиной CADASIL (церебральной аутосомно-доминантной артериопатии и инсульта), мутации Notch лигандов (Dll3) вызывают аутосомно-рецессивные спондилокостальные дизостозы. Не отмечено четкой связи генотипа с фенотипом. Часть носителей мутаций, выявленных в семье пробанда, несмотря на доминантный тип наследования, не имеют клинических и биохимических отклонений.
Дифференциальный диагноз:
•внепеченочная гипоплазия/атрезия;
•острые гепатиты и холангиты вирусной этиологии, аутоиммунных гепатитов;
•другие формы дуктулярной гипоплазии;
253

Атлас редких болезней
• нарушение синтеза и экскреции желчных кислот (болезнью Байлера);
• митохондральная гепатопатия;
• тирозинемия, чаще 1б типа.
|
Лечение. Показано хирургическое лечение, в боль- |
|
|
шей степени кардиохирургическое, симптоматичес- |
|
|
кое (урсодеоксихолевая кислота, коррекция витамин- |
|
|
ной недостаточности, при выраженном холестазе |
|
|
с зудом — колестирамин, седативные препараты), кор- |
|
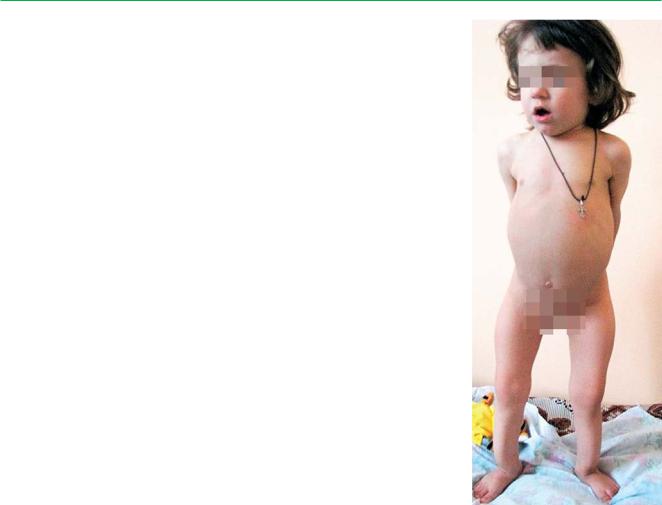
Челюстно-лицевой дисморфизм |
рекция проявлений тубулопатии (синдрома Фанкони) |
|
у больной П. 4 лет с синдромом |
в случае ее развития. Трансплантация печени нужна |
|
Алажилля (треугольное |
примерно 1/3 больных. |
|
асимметричное лицо, |
|
|
прогнатия, седловидная |
Прогноз. Причиной смерти большинства (50–75%) |
|
переносица, антимонголоидный |
||
больныхссиндромомАлажилляявляетсяпрогрессиру- |
||
разрез глаз, короткий фильтр) |
||
|
ющая сердечная недостаточность. Прогрессирование |
|
|
печеночной недостаточности определяется возрас- |
том дебюта: так, без трансплантации печени 10-летняя выживаемость отмечена у 45% больных с дебютом холестаза в неонатальном периоде и у 79% детей с поздним дебютом (средний возраст 5 лет).
Список рекомендованной литературы
1.Moyer V., Freese D.K., Whitington P.F., Olson A.D. et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2004; 39 (2): 115–128.
2.Lykavieris P., Hadchouel M., Chardot C., Bernard O. Outcome of liver disease in children with Alagille syndrome: a stadu of 163 patients. Gut. 2001; 49: 431–435.
3.Kamath B.M., Bason L., Piccoli D.A. et al. Consequenes og JAG1 mutation. J Med Genet. 2003; 40: 891–895.
4.McDaniell R., Warthen D., Sanchez-Lara P., Pai A. et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of Notch signaling pathway. The Am J of Human Genetics. 2006; 79: 169–173.
5.Krantz I., Piccoli D., Spinner N. Alagille syndrome. J Med Genet. 1997; 34: 152–157.
6.Martin S., Garel L., Alvares F. Alagille’s syndrome associated with cystic renal disease.
Arch of Disease in Childhood. 1996; 74: 232–235.
7.Jones E., Clement-Jones M., Wilson D. JAGGED1 expression in human embryos: correlation with the Alagille syndrome phenotype. J Med Genet. 2000; 37: 658–662.
254
СИНДРОМ АНГЕЛЬМАНА
(ANGELMAN SYNDROME)
МКБ-10: Q93.5; OMIM 105830
Определение. Синдром Ангельмана — это нейрогенетическое заболевание, характеризующееся задержкой интеллектуального и физического развития, нарушениями сна, приступами судорог, резкими движениями (особенно рук), частым беспричинным смехом или улыбкой и, как правило, эти люди, выглядят очень счастливыми.
В 1965 г. Гарри Ангельман описал трех мальчиков, которых наблюдал, из разных семей с тяжелой умственной отсталостью, дерганной марионеточной походкой, радостным лицом
иприступами немотивированного смеха. В 1982 г. Чарльз Уильямс
иДжейми Фриас описали ход болезни, для которой характерны задержка психического развития, нарушения сна, припадки, хаотические движения (особенно рук), частый смех или улыбки.
Синонимы: синдром веселой марионетки.
Эпидемиология. Частота встречаемости — 1:10 000–20 000 живорожденных младенцев. Соотношение полов: M1: Ж1.
Этиология, патогенез. Существуют несколько генетических механизмов синдрома, и все они связаны с хромосомным сегментом 15q11–q13:
•вновь возникшие интерстициальные делеции 15-й хромосомы, доставшейся от матери (70–75%);
•отцовская однородительская дисомия по 15-й хромосоме (2%);
•мутация гена UBE3A, кодирующего убиквитин-протеин-лигазу (5–10%).
В остальных случаях точная причи-
на болезни остается неизвестной. |
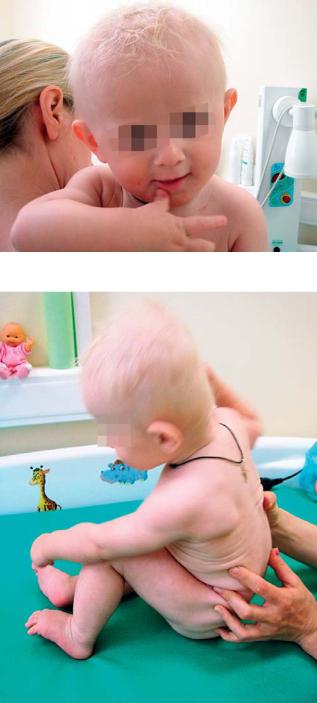
Внешний вид больного с синдромом Ангельмана |
255

Атлас редких болезней
Клинические проявления
Для синдрома Ангельмана характерны:
• задержка психомоторного развития: тяжелая умственная отсталость с выраженной задержкой двигательного развития (100%), приступы немотивированного смеха, отсутствие речи или крайне малый набор слов — не более 6 (100%);
•фенотип: микробрахицефалия, светлые волосы (65%), глубоко посаженные глаза; аномалии глаз (в том числе гипопигментация сосудистой оболочки и радужки — у 88% больных светло-голубые глаза); косоглазие (42%); верхняя микрогнотия; большой рот с высунутым языком и широкими межзубными промежутками; нижняя прогнатия;
•со стороны центральной нервной системы: атаксия, частые хаотичные движения руками, подергивание рук при ходьбе, напоминающие движения марионетки (100%); эпилептические припадки, от больших до акинетических, чаще начинающиеся в 18–24 мес (86%); мышечная гипотония; иногда гиперрефлексия, леворукость.
Рентгенологическая картина. При КТ и МРТ: атрофия больших полушарий.
Диагноз основывается на клинических критериях. Для подтверждения диагноза проводится молекулярно-генетическое исследование.
Лечение. Синдром Ангельмана является врожденной генетической аномалией; в настоящее время специфические способы его лечения не разработаны. Однако, некоторые лечебные мероприятия повышают качество жизни людей с синдромом. В частности, младенцы с гипотонусом должны получать массаж и другие виды специальной терапии (физиотерапии). Рекомендуется использование специальных методик развития ребенка, занятия с логопедом и дефектологом. Нарушения сна корректируются назначением легких снотворных, эпилептические приступы — приемом противосудорожных препаратов; нарушения стула регулируются назначением легких слабительных.
Рекомендуется проведение ЭЭГ, ультразвукового исследования органов брюшной полости 1 раз в 3 мес, регулярное определение уровня печеночных ферментов.
Прогноз. Рекомендуется обучать таких детей языку жестов. Занятия с раннего возраста по специальным программам, направленные на развитие навыков мелкой и общей моторики, в ряде случаев дают хорошие результаты. Перспективы развития зависят от степени пораженности хромосомы. Некоторые люди с синдромом Ангельмана способны освоить навыки самообслуживания и речь на примитивном уровне (обычно причиной синдрома в этом случае становится мутация), другие никогда не смогут ходить и говорить (это обычно происходит
вслучае делеции части хромосомы). С возрастом, как правило, симптомы гиперактивности и нарушения сна смягчаются. У девочек с синдромом Ангельмана
впериод полового созревания могут участиться припадки. Большинство людей с синдромом Ангельмана способны контролировать экскреторные функции (мочеиспускание и дефекацию) днем, немногие — и ночью. Некоторые люди
256

Синдром Ангельмана
с синдромом Ангельмана способны есть при помощи ножа и вилки, одеваться самостоятельно в случае отсутствия на одежде пуговиц, «молний». Во взрослом возрасте может появиться ожирение и ухудшиться состояние позвоночника (сколиоз). Менструации, половое созревание индивидов с синдромом Ангельмана происходит в обычные сроки.
Клинический пример
Мальчик 8 мес. Жалобы на задержку психомоторного развития.
Из анамнеза: ребенок с рождения наблюдался с диагнозом «Задержка психомоторного развития», проводились курсы массажа, ноотропная терапия. Семейный анамнез не отягощен.
При осмотре: уплощение затылка, расходящееся косоглазие, широкий рот, высунутый наружу язык, выдающийся вперед подбородок. синдром диффузной мышечной гипотонии, задержка моторного и психо-речевого развития, избыточная хаотичная двигательная активность (см. рис. на стр. 255).
ЭЭГ: эпилептиформная активность, Консультирован генетиком: заподозрен синдром Ангельмана.
Генетический анализ на синдром Ангельмана — подтвержден. Лечение: симптоматическое, согласно клинической картине. Наблюдение неврологом, эндокринологом, офтальмологом.
Список рекомендованной литературы
1.Yamasaki K., Joh K., Ohta T., Masuzaki H., Ishimaru T., Mukai T., Niikawa N., Ogawa M., Wagstaff J., Kishino T. Neurons but not glial cells show reciprocal imprinting of sense and antisense transcripts of Ube3a. Hum Mol Genet. 2003; 12: 837–847.
2.Angelman H. «Puppet» children: A report on three cases. Dev Med Child Neurol. 1965; 7: 681.
3.Clayton-Smith J., Loan L. Angelman syndrome: A review of the clinical and genetic aspects. J Med Genet. 2003; 40: 87.
4.Hall B.D. Adjunct diagnostic test for Angelman syndrome: The tuning fork response. Am J Med Genet. 2002; 109: 238.
257
СИНДРОМ БАРТТЕРА
(BARTTER SYNDROME)
МКБ-10: E26.8; OMIM 601678, 607364, 241200, 602522
Определение. Синдром Барттера — редкое генетическое заболевание, обусловленное дефектом реабсорбции Na и Cl в толстом восходящем колене петли Генле, для которого характерно развитие гипокалиемии, гипохлоремии и метаболического алкалоза.
Синонимы: нормотензивный гиперальдостеронизм.
Эпидемиология. Распространенность составляет 1 на 830000 населения.
Тип наследования: аутосомно-рецессивный. Существует несколько вариантов синдрома Барттера.
Этиология, патогенез. В зависимости от мутации, вызвавшей гипокалиемию, выделяют несколько вариантов синдрома Барттера.
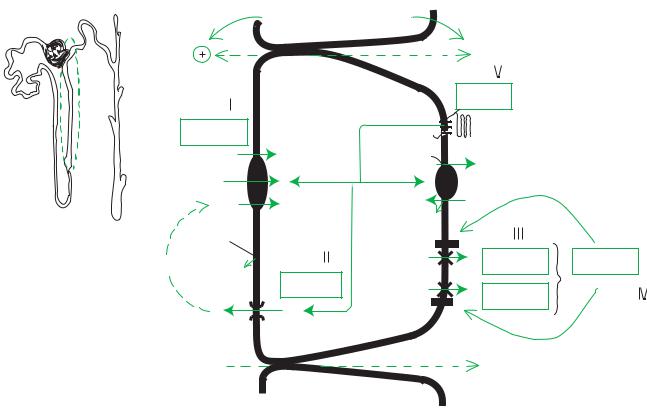
•Неонатальный вариант, тип 1 — мутация гена, расположенного на хромосоме 15 (q15–21), с локализацией SLC12A1. Этот ген кодирует почечный котранспортер Na+–K+–2Cl–, расположенный в толстом восходящем отделе петли Генле.
|
|
|
TAL cell |
|
|
|
|
Luminal |
|
Lumen/positive voltage |
Basolateral |
||
|
|
|
|
|||
|
spase |
|
|
|
spase |
|
|
|
|
|
|
Type |
|
TAL |
Type |
|
Inhibitory |
CaSR |
|
|
|
|
|
|
|||
|
|
|
|
Ca2+ |
|
|
|
NKCC2 |
|
|
|
||
|
|
|
|
|
||
|
Na+ |
|
ATP |
|
|
|
|
2Cl/ |
|
|
3Na+ |
Na+, K+/ATPase |
|
|
|
|
|
|||
|
K |
+ |
|
|
2K+ |
|
|
|
|
|
|
|
|
|
H2O |
|
Cl/ |
Type |
|
|
|
Type |
|
|
|||
K recycling |
|
CIC/Kb |
Barttin |
|||
|
|
|
RCMK |
Cl |
CIC/Ka |
Type |
|
|
|
K+ |
|||
|
|
|
|
|
|
|
[Na+, Ca2+, Mg2+] |
|
|
|
|
|
|
Локализация и механизмы нарушений при различных вариантах синдрома Барттера
258
Синдром Барттера
• |
Неонатальный вариант, тип 2 — |
|
мутация гена, кодирующего АТФ- |
|
чувствительный внутренний рек- |
|
тифицирующий калиевый канал |
|
(ROMK) на хромосоме 11 (q24-25). |
• |
Классический вариант, тип 3 — |
|
дефектный ген кодирует почечный |
|
хлоридный канал (CLCNKB), осущест- |
|
вляющий обратный вольтаж-зави- |
|
симый транспорт хлора через базо- |
|
латеральную мембрану в кровоток. |
• Синдром Барттера с нейросенсор- |
|
|
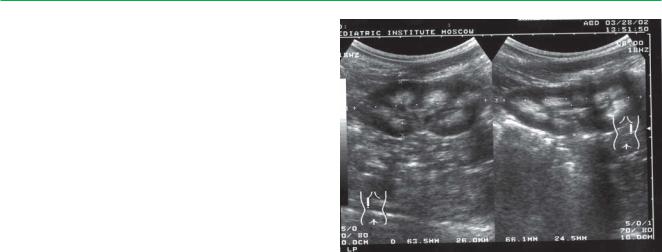
ной тугоухостью, тип 4 — мутация Ультразвуковое исследование почек. |
гена, кодирующего белок барттин |
Нефрокальциноз у ребенка в возрасте 1 года |
|
|
(BSND), который является субъеди- |
|
ницей хлоридного канала (CLCNKA и CLCNKB). |
|
•Синдром Барттера, ассоциированный с аутосомно-доминантной гипокальциемией — мутация гена кальций-чувствительного рецептора (CaSR).
При типе 1 неонатального варианта первичный дефект Na+–K+–2Cl–-
котранспортера приводит к нарушению реабсорбции натрия в толстом восходящем колене петли Генле. Потеря натрия ведет к снижению внутрисосудистого объема, активации продукции ренина и альдостерона, гиперкалиурии с последующей гипокалиемией и метаболическим алкалозом.
При типе 2 неонатального варианта нарушение функции канала ROMK препятствует возвращению реабсорбированного калия в просвет толстого восходящего колена петли Генле, что снижает функцию Na+–K+–2Cl–-котранспортера.
При неонатальном варианте синдрома Барттера развивается гиперкальциурия и нефрокальциноз.
Классический вариант сопровождается нарушением реабсорбции хлорида натрия на люминальной мембране, что ведет к гиповолемии и последующей активации ренин-ангиотензиновой системы с развитием гипокалиемического метаболического алкалоза; нефрокальциноз не развивается.
Клинические проявления (общие для всех вариантов)
•Антенатальный период: полигидроамнион (многоводие).
•Постнатальный период: низкая масса тела при рождении, гипо-, адинамия, выраженная полиурия с эпизодами тяжелой дегидратации, лихорадка, судороги, диарея.
•При типе 4 уже на 1-м месяце жизни развивается нейросенсорная тугоухость.
•Нарушения кислотно-щелочного состояния и содержания электролитов: метаболический алкалоз, гипокалиемия, гиперкальциурия, гипернатриемия, гипомагниемия, повышение уровня ренина и альдостерона.
Диагностика
Характерная клиническая симптоматика Лабораторная диагностика:
•кислотно-щелочное состояние — метаболический алкалоз;
259