

5 курс / Госпитальная педиатрия / Атлас_редких_болезней_Педиатрия,_2016_г_
.pdfАтлас редких болезней
•биохимический анализ крови — гипокалиемия, гиперкальциурия, гипернатриемия, гипомагниемия, повышение уровня ренина и альдостерона, простагландина Е2;
•общий анализ мочи — гиперкальциурия, гипостенурия;
•ультразвуковое исследование почек — нефрокальциноз.
Важным диагностическим признаком является нормальное артериальное
давление (отсутствие артериальной гипертензии, несмотря на гиперрениновое состояние).
При классическом варианте синдрома Барттера гиперкальциурия и нефрокальциноз отсутствуют.
Дифференциальный диагноз:
•случаи гипокалиемического алкалоза, обусловленного избыточным применением петлевых или тиазидных диуретиков, рвотой, кишечной потерей натрия и калия.
Лечение. В неонатальном периоде необходимо устранение дегидратации путем продолжительных инфузий изотонического раствора хлорида натрия и восполнения недостатка хлорида калия.
При неонатальных вариантах болезни для купирования гипокалиемии и метаболического алкалоза без последующего введения хлорида калия эффективно лечение индометацином (не ранее 4–6-й недели жизни) в суточной дозе не более 1 мг/кг.
Основа терапии классического варианта — индометацин в дозе 2–5 мг/кг в сутки. Кроме того, необходимо восполнение потерь калия путем назначения калия хлорида, иногда в сочетании со спиронолактоном. Лечение назначают пожизненно.
Прогноз. При неонатальных вариантах — неблагоприятный. Терапия индометацином приводит к коррекции электролитного баланса, но гиперкальциурия сохраняется, нефрокальциноз медленно прогрессирует, приводя к почечной недостаточности.
При классическом варианте прогноз благоприятный в случае адекватной коррекции алкалоза и электролитных нарушений.
Клинический пример
Девочка, возраст 10 мес. Поступила в нефрологическое отделение НЦЗД впервые с направительным диагнозом «Тубулопатия».
Из анамнеза. Ребенок родился недоношенным. Сразу после рождения диагностирована динамическая кишечная непроходимость. С 5-го мес жизни стали появляться и прогрессировать вялость, сонливость, мышечная гипотония, снижение аппетита, отставание в физическом и психомоторном развитии. В возрасте 10 мес в тяжелом состоянии поступила в стационар по месту жительства, где выявлена выраженная гипокалиемия (1,4–2,7 ммоль/л), рефрактерная к инфузиям хлористого калия (KCl).
При осмотре в отделении: резкое отставание в физическом и психомоторном развитии, выраженная мышечная гипотония, АД в норме.
Со стороны внутренних органов: физикальные отклонения не отмечались.
260

Синдром Барттера
По лабораторным данным: тяжелый метаболический алкалоз (pH 7,612, SB 47,2 ммоль/л), выраженная гипонатриемия (119 ммоль/л), гипокалиемия (2,3 ммоль/л), гипохлоремия (67 ммоль/л).
По данным УЗИ почек: нефрокальциноз не определяется. Диагноз. Синдром Бартера, классический вариант.
Лечение. Назначен индометацин, перорально раствор калия хлорида 4% под контролем кислотно-щелочного состояния и уровня электролитов крови.
Результаты. На фоне проводимой терапии в течение 9 мес отмечена положительная динамика: уменьшение метаболического алкалоза, нормализация уровней калия, натрия и хлоридов; девочка выросла на 12 см, прибавила в весе 2 кг; уменьшилась выраженность мышечной гипотонии.
Список рекомендованной литературы
1.Зверев Я.Ф., Брюханов В.Н., Лампатов В.В. Нефрология. 2004; 8 (4): 11–21.
2.Симптомы и синдромы в эндокринологии. Справочное пособие. Под ред. Ю.И. Караченцева. 1-е изд. Харьков: ООО «СА.М.». 2006. С. 31. ISBN 978-966-8591- 14-3.
3.Bartter F.C. et al. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis: a new syndrome. Am J Med. 1962; 33: 811–828.
4.Ji W., Foo J.N., O’Roak B.J. et al. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat Genet. 2008; 40: 592.
5.Seyberth H.W. An improved terminology and classification of Bartter-like syndromes.
Nat Clin Pract Nephrol. 2008 Aug.
6.Simon D.B. et al. Mutations in the gene CLCNKB cause Bartter’s syndrome type III. Nat Genet. 1997; 17: 171–178.
7.Proesmans W. Threading through the mizmaze of Bartter syndrome. Pediatr Nephrol. 2006; 21: 896.
8.Tourne G., Collet F., Varlet M.N. et al. Prenatal Bartter’s syndrome. Report of two cases.
J Gynecol Obstet Biol Reprod (Paris). 2003; 32: 751.
261

СИНДРОМ БЕКВИТА–ВИДЕМАНА
(BECKWITH-WIEDEMAN SYNDROME)
МКБ-10: Q87.3; ОМIМ 130650
Определение. Синдром Беквита–Видемана — комплекс врожденных аномалий, который характеризуется сочетанием макроглоссии, омфалоцеле и другими пупочными аномалиями, а также аномалиями (висцеромегалией) почек, надпочечников, поджелудочной железы, яичек. Быстрый рост сопровождается ускоренным развитием костей; типична гипогликемия; отмечается склонность к образованию злокачественных опухолей (нефробластома или опухоль Вильмса).
Синдром Беквита–Видемана впервые был описан J.В. Beckwith в 1963 г. и H.R. Wiedemann в 1964 г.
Синонимы: синдром экзомфалии–макроглоссии–гигантизма.
Эпидемиология. 1 на 13700 новорожденных. Соотношение полов: M1: Ж1.
Тип наследования: большинство случаев изолированные. Описаны случаи с аутосомно-доминантным типом наследования с варьирующей экспрессивностью.
Этиология, патогенез. В большинстве случаев синдром Беквита–Видемана возникает спорадически. Обнаруживают структурные и функциональные аномалии критического района короткого плеча хромосомы 11. Гены CDKN1C и IGF2 расположены в регионе 11p 15.5. При семейных случаях почти в 40% выявляют мутации гена CDKN1C, а в спорадических — не более 5%. В гене, экспрессирующимся с отцовской хромосомы, мутации не выявляются, но возможна диаллельная экспрессия гена с материнской хромосомы.
Клинические проявления
Выделяют большие и малые клинические критерии.
Внешний вид больного с синдромом Беквита– Ведемана
Большие клинические признаки
•макроглосия у 95% пациентов;
•макросомия, или гигантизм, у 80% пациентов; склонность к чрезмерному росту в основном проявляется в детстве, с возрастом становится менее выраженной;
•дефекты брюшной стенки; у 65% пациентов имеется пупочная грыжа, диастаз прямых мышц живота;
•органомегалия — увеличение размеров почек, печени, селезенки, поджелудочной железы и надпочечников у 50% пациентов.
262

Синдром Беквита–Видемана
Малые клинические признаки
•гипогликемия в неонатальный период встречается у 65% пациентов, в основном преходящая и слабо выраженная;
•аномалии почек: дефекты, дисплазия мозгового вещества;
•небольшая складка на мочке уха у 30% пациентов;
•«пылающий» (винный) невус на лице у 30% пациентов;
•гемигиперплазия — увеличение одной половины тела у 30–35% пациентов;
•эмбриональные опухоли: нейробластома, опухоль Вильмса, карцинома надпочечников, гепатобластома, рабдомиосаркома выявляются у 7,5% пациентов;
•многоводие (при беременности у матери).
Рентгенологическая картина. Костный возраст опережает паспортный, на рентгенограмме отмечается расширение метафизов и сужение диафизов длинных трубчатых костей. Микроцефалия или гидроцефалия; гипоплазия верхней челюсти и гиперплазия нижней. Висцеромегалия. Может быть диафрагмальная грыжа, аномалии долевого строения легких, незаконченный поворот кишечника, дефект межжелудочковой перегородки. Склонность к образованию злокачественных опухолей.
Диагностика основывается на клинических критериях. Для постановки диагноза достаточно 2 больших и одного малого критерия. Для подтверждения диагноза проводится молекулярно-генетическое исследование.
Дифференциальный диагноз:
•гипотиреоз врожденный;
•омфалоцеле;
•семейная врожденная гипогликемия.
Лечение. Некорригированная неонатальная гипогликемия является серьезным осложнением, что может повлечь за собой дальнейшие церебральные дисфункции, такие как судороги, умственная отсталость (от слабой до умеренно выраженной), в более тяжелых случаях способна стать причиной неонатальной гибели. Макроглоссия вызывает различные патологические состояния: от затруднений при вскармливании до обструкции дыхательных путей и гибели. Отдаленные осложнения включают высокий риск развития опухолей органов брюшной полости, в особенности опухоли Вильмса, гепатобластомы, нейробластомы и злокачественной опухоли коры надпочечника.
Рекомендуется эхографическое обследование ребенка каждые три месяца в течение первых 6 лет жизни для выявления опухолей органов брюшной полости; регулярное определение уровня глюкозы крови, определение альфафетопротеина как маркера гепатобластомы. Удаление пупочной грыжи и других дефектов брюшной стенки.
Прогноз. Уровень неонатальной смертности составляет примерно 21% . Для выживших прогноз в целом бывает благоприятным и зависит от тяжести сочетанных аномалий и наличия отдаленных осложнений.
263

Атлас редких болезней
Список рекомендованной литературы
1.DeBaun M.R., Niemitz E.L., McNeil D.E., Brandenburg S.A., Lee M.P., Feinberg A.P. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith– Wiedemann syndrome with cancer and birth defects. Am J Hum Genet. 2002;
70:604–611.
2.Rump P., Zeegers M.P., van Essen A.J. Tumor risk in Beckwith–Wiedemann syndrome: a review and meta-analysis. Am J Med Genet. 2005; 136: 95–104.
3.Goldman M., Smith A., Shuman C. et al. Renal abnormalities in Beckwith–Wiedemann syndrome are associated with 11p15.5 uniparental disomy. J Am Soc Nephrol. 2002;
13:2077–2084.
4.Cooper W.N., Luharia A., Evans G.A. et al. Molecular subtypes and phenotypic expression of Beckwith–Wiedemann syndrome. Eur J Hum Genet. 2005; 13: 1025–1032.
5.Weksberg R., Shuman C., Smith A.C. Beckwith–Wiedemann Syndrome. Am J of Med Genet. 2005; 137: 12–23.
6.Enklaar T., Zabel B.U., Prawitt D. Beckwith–Wiedemann syndrome: multiple molecular mechanisms. Expert Rev Mol Med. 2006; 8: 1–19.
264
СИНДРОМ БЛОХА–СУЛЬЦБЕРГЕРА
(BLOCH–SULZBERGER SYNDROME)
МКБ-10: Q82.3; ОМIМ 308300
Определение. Редкий нейроэктодермальный мультисистемный генодерматоз, характеризующийся своеобразным поражением кожи и ее придатков (волос и ногтей) в сочетании с патологией центральной нервной системы, а также глаз, зубов, скелета, реже — других органов. Впервые описано Bloch в 1926 г. и Sulzberger в 1927 г.
Синонимы: недержание пигмента, синдром Асбо-Хансена, линейный меланобластоз, дегенеративный меланоз Сименса, пигментный дерматоз Сиренс-Блох.
Эпидемиология. Частота встречаемости составляет 0,7 на 100 000 новорожденных.
Тип наследования: Х-сцепленный доминантный (в большинстве случаев). Заболевание летально для плодов мужского пола на ранних стадиях внутриутробного развития. Описаны случаи рождения лиц мужского пола с синдромом Блоха–Сульцбергера, их выживаемость обусловливалась либо спонтанными мутациями, либо сопутствующими заболеваниями, такими как дисомия по Х-хромосоме (синдром Клайнфельтера) или соматический мозаицизм мутантной хромосомы Х.
Этиология, патогенез. Патология возникает в результате нестабильности хромосом и развивается вследствие врожденной мутации гена IKBKG (inhibitor of kappa b kinase gamma) и его продукта NEMO (nuclear factor-kB essential modulator)/IKK (inhibitor kappa kinase- ), играющего роль в активации NF-kB. Мутации гена IKBKG локализованы в Х-хромосоме (локус Xq28).
В патогенезе основную роль играет наличие популяции дефицитных по NEMO клеток, что приводит к апоптозу (клинически проявляется воспалением и образованием везикулезных элементов); затем уменьшение количества дефицитных по NEMO клеток и пролиферация неизмененных NEMO-кератиноцитов приводит к формированию веррукозных папул. Проявление гиперпигментации обусловлено проникновением из эпидермиса в дерму меланинового пигмента.
Клинические проявления
Первые признаки отмечаются при рождении или в первые дни, а иногда
инедели. Выделяют 4 стадии клинических проявлений, сменяющих последовательно друг друга.
1-я стадия — воспалительная (везикуло-буллезная). Характеризуется появлением на эритематозно-отечном фоне пузырей и пузырьков, редко — уртикарий
ипустул. Пузырные элементы имеют плотную покрышку с прозрачным содержимым, локализуются на лице, туловище, верхних и нижних конечностях и располагаются линейно. После вскрытия элементов формируются эрозии, покрытые серозными корочками.
265

Атлас редких болезней
2-я стадия — папуло-веррукозная. На 5–10-й нед жизни появляются плотные лентикулярные папулы, локализующиеся на тыльной поверхности верхних и нижних конечностей и располагающиеся линейно, напоминая бородавчатый невус. Одновременно развивается диффузный гиперкератоз. Стадия продолжается до нескольких месяцев.
3-я стадия — гиперпигментная. Развивается к концу первого полугодия жизни ребенка и характеризуется гиперпигментными пятнами темно-корич- невого, кофейного или темно-серого цвета со светлыми краями. Высыпания имеют вид параллельно идущих полос с неправильными V-образными и зигзагообразными краями. Пигментные пятна имеют причудливый вид, напоми-
нают «брызги грязи», «искры фейерверка», «завитки», «спирали», «кольца» и т. д. Характерно расположение по линиям Блашко в виде «мраморного кекса». Гиперпигментированные очаги обычно сохраняются до 5–6 лет, но могут оставаться до 30-летнего возраста, после чего, по некоторым данным, исчезают спонтанно.
4-я стадия — атрофо-гипопигментная. Развивается не у всех пациентов. Образуются очаги линейной атрофии и гипоили депигментации. В участках гипопигментации отсутствуют придатки кожи.
Помимо поражения кожи характерны и другие (внекожные) проявления: поражения и/или аномалии развития зубов (дефицит дентина, диастема), глаз (страбизм, псевдоглиома, нистагм, кератиты, катаракта, косоглазие; отслойка сетчатки, которая может возникнуть на первом году жизни; атрофия зрительного нерва), центральной нервной системы (олигофрения, парезы, спастические параличи, эпилепсия, микроцефалия, атаксия), волос (алопеция, редкие волосы, аномалии бровей и ресниц), ногтей (дистрофия), сосков (добавочный) и молочных желез, черепа, неба, челюстей. К редким внекожным изменениям относятся пороки сердца, врожденные вывихи бедра, асимметрия грудной клетки.
Диагностика
Диагноз выставляется на основании клинической картины и характерных патогистологических признаков, соответствующих каждой стадии заболевания.
Для постановки диагноза достаточно наличия одного основного критерия, т. е. проявления одной из четырех стадий заболевания.
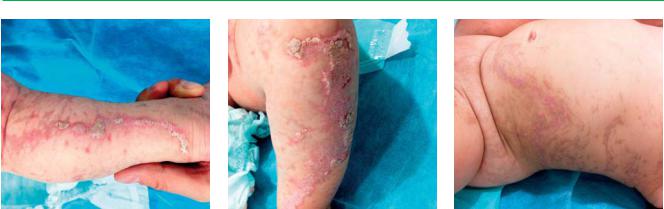
Синдром Блоха–Сульцбергера
А |
Б |
В |
Ребенок в возрасте 3 мес: |
Высыпания |
3-я стадия клинических |
линейные папуло-веррукозные |
на разгибательной |
проявлений — формирование |
высыпания, на поверхности |
поверхности левой голени. |
гиперпигментых пятен |
которых отмечаются |
Отмечается диффузный |
красновато-бурого, местами |
гиперкератотические наслоения. |
гиперкератоз |
коричневатого цвета |
Также отмечается формирование |
|
причудливых форм в виде |
гиперпигментных пятен |
|
«завитков» и «брызг грязи». |
красновато-бурого цвета |
|
Высыпания расположены |
на местах бывших эрозий |
|
по линиям Блашко |
Дополнительные критерии (внекожные поражения) необязательны для подтверждения диагноза, но могут проявляться.
Дифференциальный диагноз. Необходимо учитывать возраст пациента и стадию генодерматоза.
Воспалительная стадия (везикуло-буллезная) у новорожденных:
•врожденный буллезный эпидермолиз;
•врожденная буллезная ихтиозоформная эритродермия;
•врожденная порфирия;
•энтеропатический акродерматит;
•потница;
•инфекционные болезни (эпидемическая пузырчатка новорожденных, эксфолиативный дерматит, буллезное импетиго, кандидоз, герпетическая инфекция, ветряная оспа);
•приобретенные неинфекционные заболевания (токсическая эритема новорожденных, транзиторный неонатальный пустулезный меланоз, детский акропустулез, эозинофильный пустулезный фолликулит).
Папуло-веррукозная стадия:
•вульгарные бородавки;
•веррукозный невус;
•контагиозный моллюск;
•линейный эпидермальный невус;
•точечная хондродисплазия, наследуемая по доминантному Х-сцепленному типу.
Стадия гиперпигментации:
•гипомеланоз Ито;
•синдром Негели–Блоха–Ядассона.
267

Атлас редких болезней
Атрофо-гипопигментная стадия:
•витилиго;
•рубцовая алопеция;
•различные формы эктодермальной дисплазии.
Лечение симптоматическое, зависит от клинических проявлений и сопутствующей патологии. Пузыри вскрывают, обрабатывают антисептическими средствами, тушируют анилиновыми красителями. На эритематозные пятна назначают 10% цинковое масло, топические глюкокортикостероиды (предпочтительно слабой степени активности).
Прогноз зависит от выраженности кожного процесса и сопутствующей патологии. При легких формах прогноз благоприятный: описаны случаи спонтанного самоизлечения. При тяжелых формах с поражением внутренних органов заболевание, как правило, прогрессирует и приводит к тяжелым осложнениям (кровотечения, инфекции, плоскоклеточный рак кожи), которые часто заканчиваются летальным исходом.
Список рекомендованной литературы
1.Bloch B. Eigentumliche bisher nichtbeschreibene Pigmentaffektion (Incontinentia Pigmenti). Schweiz med Wchnschr. 1926; 56: 404.
2.Sulzberger M.B. Uber eine bisher nicht beschriebene congenitale Pigmentanomalie.
Arch Dermat Syph. 1928; 154: 19.
3.Orphanet Report Series — Prevalence of rare diseases: Bibliographic data May 2014, № 1. URL: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_ diseases_by_alphabetical_list.pdf
4.Genomic rearrangement in NEMO impairs NF-kappa-B activation and is a cause of incontinentia pigmenti. International Incontinentia Pigmenti Consortium Nature.
2000; 405 (6785): 466–72.
5.Miniс S. et al. Incontinentia pigmenti diagnostic criteria update. Clin Genet. 2014; 85 (6): 536–542.
6.Дерматология Фицпатрика в клинической практике. К. Вольф, Лоуэлл А. Голдсмит, Стивен И. Кац, Барбара А. Джилкрест, Эми С. Пааер, Дэвид Дж. Леффель. Пер. с англ. В.П. Адаскевича, М.В. Гатмана. М.: Бином. 2012. С. 689–690.
7.Hadj-Rabia S., Rimella A., Smahi A. et al. Clinical and histologic features of incontinentia pigmenti in adults with nuclear factor-kB essential modulator gene mutations. J Am Acad Dermatol. 2011; 64: 508–515.
268

СИНДРОМ КОСТЕЛЛО
(COSTELLO SYNDROME)
МКБ-10: Q80–Q89; ОМIМ 218040
Определение. Синдром Костелло — редкое заболевание, характеризующееся множественными врожденными аномалиями: постнатальной задержкой роста, грубыми чертами лица, кожными изменениями, диффузной гипотонией и сердечной патологией (гипертрофическая кардиомиопатия, врожденные пороки сердца, аритмия). Отмечается предрасположенность к развитию опухолей.
Впервые описан в 1977 г. доктором Джеком Костелло (Jack Costello) из Новой Зеландии. Возникновение заболевания связывают с мутацией гена HRAS на коротком плече 15-й хромосомы.
Синонимы: гипогиперпаратиреоидизм, диспаратиреоидизм, Костелло–Дента (Costello–Dent) синдром, псевдо-псевдогипопаратиреоидизм.
Эпидемиология. Встречаемость: 1:300000 в Великобритании, 1:24000000 человек в Японии. В мире зарегистрировано около 300 пациентов с данным синдромом. Одинаково часто встречается среди женщин и мужчин.
Тип наследования: аутосомно-доминантный; часто спорадические случаи.
Этиология, патогенез. Возникновение заболевания связывают с мутацией гена HRAS на коротком плече 15-й хромосомы (11p15.5), его локализация — в 12-м или 13-м кодонах.
HRAS — известный онкоген; его аномальная активность часто встречается в случаях спорадических соматических опухолей; обусловливает повышенную заболеваемость раком легких и мочевого пузыря у лиц с наличием мутаций в этом гене. Данный ген кодирует синтез сверхактивного белка HRAS, который вызывает непрерывное сверхактивное клеточное деление и рост клеток.
Ген HRAS состоит из 6 экзонов. Наиболее частые мутации, приводящие к развитию синдрома Костелло, локализованы в экзоне 2. В 90% случаев это 12-й или 13-й кодоны гена HRAS. Наиболее частые
мутации — р.G12 и р.G13. Выявлены корреляции между фенотипом больных синдромом Костелло и разновидностью мутаций в гене HRAS. Так, наиболее часто злокачественные опухоли обнаруживались у пациентов с мутацией р.Gly12Ala (57%). В то же время ни у одного из пациентов с мутацией р.Gly13Cys не выявлено злокачественных опухолей, мультифокальной предсердной тахикардии и роста папиллом.
269