

5 курс / Госпитальная педиатрия / Атлас_редких_болезней_Педиатрия,_2016_г_
.pdfАтлас редких болезней
2.Burwinkel B., Maichele A. J., Aagenaes O., Bakker H.D., Lerner A., Shin Y.S., Strachan J.A., Kilimann M.W. Autosomal glycogenosis of liver and muscle due to phosphorylase kinase deficiency is caused by mutations in the phosphorylase kinase beta subunit (PHKB). Hum. Molec. Genet. 1997. 6: 1109–1115.
3.Ohtani Y., Matsuda I., Iwamasa T., Tamari H., Origuchi Y., Miike T. Infantile glycogen storage myopathy in a girl with phosphorylase kinase deficiency. Neurology. 1982. 32: 833–838.
4.Ozen H. Glycogen storage diseases: new perspectives . World J. Gastroenterol. 2007; 13 (18): 2541–2553.
120

Гликогеновая болезнь
ГЛИКОГЕНОВАЯ БОЛЕЗНЬ IXc ТИПА
(GLYCOGEN STORAGE DISEASE TYPE IХс)
МКБ-10: Е74.0; OMIM 613027, 172471
Определение. Гликогеновая болезнь IXc типа — аутосомно-рецессивное заболевание, вызываемое дефицитом тестикулярной/печеночной изоформы-субъединицы киназы фосфорилазы и ассоциированное накоплением гликогена в печени и мышцах.
Синонимы: недостаточность тестикулярной/печеночной изоформы-субъединицы киназы фосфорилазы.
Эпидемиология. Имеются единичные описания.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Мутации гена PHKG2, кодирующего тестикулярную/ печеночную изоформу -субъединицы киназы фосфорилазы.
Ген картирован на 16р11.2. Установлено, что ген PHKG2 содержит 10 экзонов и занимает 9,5 kb геномной ДНК.
Патогенез аналогичен таковому при IXa подтипе.
Клинические проявления
Клиническая симптоматика в целом сходна с таковой при VI типе гликогеновой болезни. В большинстве случаев со временем эти симптомы уменьшаются, однако, у некоторых имеется высокий риск развития фиброза и цирроза печени.
Диагностика
Клиническая симптоматика Лабораторная диагностика
•Кровь: гипогликемия и кетоацидоз натощак, лактат-ацидоз, гипертриглицеридемия.
Морфологическая диагностика
•Гистологические изменения: фиброз печени различной степени выраженности вплоть до формирования цирроза, пролиферация желчных протоков.
•Гистохимическое исследование: снижение активности киназы фосфорилазы в печени и мышцах.
Лечение. В зависимости от тяжести течения болезни лечение либо не проводится, либо оно аналогично терапии гликогеновой болезни VI типа.
121

Атлас редких болезней
Список рекомендованной литературы
1.Beauchamp N. J., Dalton A., Ramaswami U., Niinikoski H., Mention K., Kenny P., Kolho K.-L., Raiman J., Walter J., Treacy E., Tanner S., Sharrard M. Glycogen storage disease type IX: high variability in clinical phenotype. Molec. Genet. Metab. 2007. 92: 88–99.
2.Burwinkel B., Tanner M.S., Kilimann M.W. Phosphorylase kinase deficient liver glycogenosis: progression to cirrhosis in infancy associated with PHKG2 mutations (H144Y and L225R). J. Med. Genet. 2000. 37: 376–377.
3.Kagalwalla A.F., Kagalwalla Y.A., al Ajaji S., Gorka W., Ali M.A. Phosphorylase b kinase deficiency glycogenosis with cirrhosis of the liver. J. Pediat. 1995. 127: 602–605.
4.Lerner A., Iancu T.C., Bashan N., Potashnik R., Moses S. A new variant of glycogen storage disease: type IXc. Am. J. Dis. Child. 1982. 136: 406–410.
5.Maichele A.J., Burwinkel B., Maire I., Sovik O., Kilimann M.W. Mutations in the testis/ liver isoform of the phosphorylase kinase gamma subunit (PHKG2) cause autosomal liver glycogenosis in the gsd rat and in humans. Nature Genet. 1996. 14: 337–340.
122
ДЕФИЦИТ ЛИЗОСОМНОЙ КИСЛОЙ ЛИПАЗЫ
(LYSOSOMAL ACID LIPASE DEFICIENCY)
МКБ-10: E75.5; OMIM 278000
Определение. Дефицит лизосомной кислой липазы — редкое наследственное прогрессирующее заболевание из группы лизосомных болезней накопления, обусловленное мутацией гена LIPА, кодирующего лизосомную кислую липазу (LAL), что приводит к снижению активности фермента, нарушению процессов гидролиза и накоплению эфиров холестерина и триглицеридов в лизосомах всех типов клеток (кроме эритроцитов); в зависимости от степени выраженности ферментативного дефицита проявляется двумя клиническими формами — ранней тяжелой (болезнь Вольмана) и более легкой (болезнь накопления эфиров холестерина); характеризуется типичными биохимическими изменениями (повышение уровня холестерина, триглицеридов, липопротеидов низкой плотности и снижение уровня липопротеидов высокой плотности), гистологическими признаками («пенистые» клетки в биоптате костного мозга, клетках селезенки и лимфоузлов) и морфологическими изменениями в печени (увеличение и вакуолизация гепатоцитов и купферовских клеток, большое число «пенистых» гистиоцитов, перипортальный фиброз и цирроз).
Эпидемиология. Вследствие редкости и недостаточной выявляемости истинная распространенность заболевания не известна. По отдельным оценкам, в европейской популяции предполагаемая частота болезни накопления эфиров холестерина составляет 1:50000, болезни Вольмана — до 1:350000. В 1956 г. А. Абрамов, Шорр (S. Schorr) и Вольман (M. Wolman) описали данное наследственное заболевание у ребенка, рожденного от близкородственного брака. Болезнь получила название в честь невролога Моше Вольмана (1914–2009). К 2013 г., по данным мировой литературы, описано до 135 случаев болезни накопления эфиров холестерина и около 50 случаев болезни Вольмана.
Тип наследования: аутосомно-рецессивный.
Этиология, патогенез. Ген кислой липазы LIPA (c альтернативным названием LAL) расположен на длинном плече хромосомы 10 (10q23.2–23.3). В настоящее время описано более 30 мутаций LIPA. Генофенотипические корреляции ограничены, однако выявление мутации с. 894G>A в гомозиготном и компаунд-гетерозиготном состоянии обычно определяет более благоприятное течение болезни. Данная мутация характерна для европейской популяции и реже встречается среди азиатов.
Клинические проявления
Клинические проявления варьируют в зависимости от полного или частичного дефицита кислой липазы.
Полное отсутствие кислой липазы (болезнь Вольмана) приводит к ранним клиническим проявлениям (в первые недели жизни или на первом году): вследствие накопления липидов в клетках стенки кишечника развивается синдром
123
Атлас редких болезней
мальабсорбции в виде рвоты, диареи. Определяется выраженная грубая задержка психомоторного и физического развития, гиперрефлексия. У ребенка при осмотре — большой выступающий живот, гепатоспленомегалия, иногда желтуха. Характерный диагностический признак — увеличение и кальцификация надпочечников с формированием надпочечниковой недостаточности. Отмечаются прогрессирующая анемия и тромбоцитопения.
При частичном отсутствии кислой липазы развивается иная клиническая форма — болезнь накопления эфиров холестерина, характеризующаяся более поздним началом (5–15 лет) и отсутствием неврологической симптоматики. Ключевые клинические проявления — гепатомегалия или гепатоспленомегалия, повышение уровня печеночных аминотрансфераз, фиброз или цирроз печени. Редко развивается кальцификация надпочечников. У пациентов со спленомегалией может развиваться анемия и/или тромбоцитопения.
Диагностика
Наличие заболевания у пациента можно заподозрить по характерной для одной из клинических форм клинической картине.
Биохимический анализ крови: повышение активности печеночных аминотрансфераз, уровня холестерина, триглицеридов, липопротеидов низкой плотности и снижение уровня липопротеидов высокой плотности.
Клинический анализ крови: анемия, тромбоцитопения (при развитии вторичного гиперспленизма).
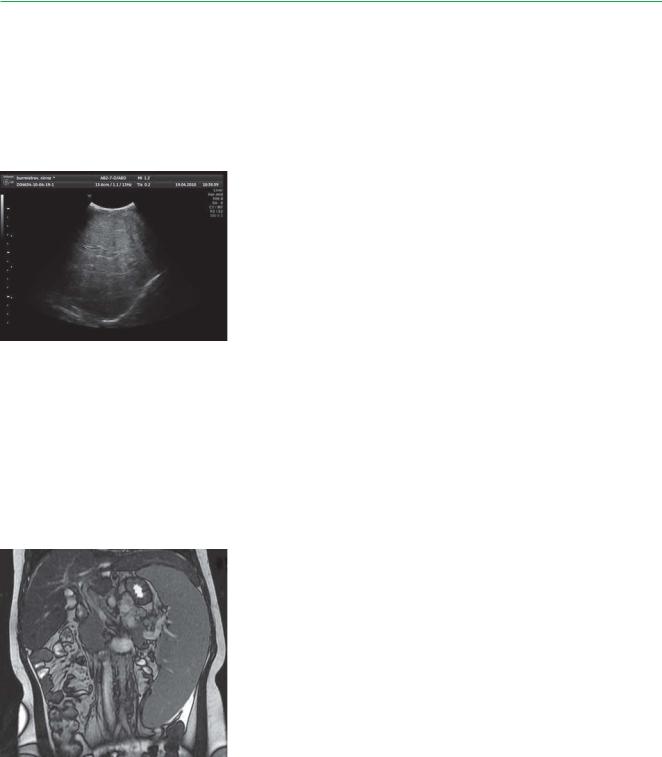
Методы визуализации (УЗИ/МРТ/КТ органов брюшной полости) помогают определить объемные и структурные изменения печени и селезенки, исключить поражение надпочечников (см. рис.).
Морфологическое исследование. При исследовании биоптата костного мозга, селезенки и лимфоузлов находят «пенистые» клетки. При описании печеночного биоптата встречаются классические признаки: увеличение и вакуолизация гепатоцитов и купферовских клеток, большое число «пенистых» гистиоцитов, перипортальный фиброз и цирроз.
«Золотым стандартом» постановки диагноза является энзимодиагностика — определение активности кислой липазы в лейкоцитах крови или сухих пятнах крови, а также молекулярно-генетическое исследование — выявление мутаций в гене LIPA.

Дефицит лизосомной кислой липазы
Лечение. Единственным патогенетическим методом лечения является длительная ферментозаместительная терапия себелипазой альфа, представляющей рекомбинантную кислую лизосомную липазу человека. В клинических исследованиях продемонстрировано улучшение морфологической картины в биоптатах печени, липидного профиля, нормализация уровня активности трансаминаз, а также увеличение выживаемости пациентов. Препарат одобрен в Европейском союзе; ожидает регистрации на территории РФ.
При отсутствии патогенетической терапии возможна симптоматическая поддержка, направленная в первую очередь на профилактику цирроза печени. В отдельных случаях положительный эффект имела трансплантация костного мозга и печени. При развитии синдрома мальабсорбции и задержки развития необходима консультация диетолога с подбором парентерального питания. Для коррекции липидного профиля возможно применение статинов и специализированная диета. При развитии надпочечниковой недостаточности назначается заместительная гормонотерапия, при выраженной анемии и тромбоцитопении — переливание крови, при выраженной спленомегалии проводят спленэктомию.
Прогноз при дефиците лизосомной кислой липазы без патогенетического лечения неблагоприятный: при болезни Вольмана летальный исход наступает на первом году жизни; продолжительность жизни при болезни накопления эфиров холестерина зависит от возраста дебюта и тяжести течения заболевания.
Клинический пример
Пациент М., возраст 6 лет 4 мес. Впервые поступил в отделение восстановительного лечения детей с болезнями пищеварительной системы НЦЗД с жалобами на слабость, вялость, быструю утомляемость, увеличение размеров живота, изменения в анализах крови.
Из анамнеза: ребенок от 1-й беременности, протекавшей на фоне отеков в III триместре. Роды самостоятельные, срочные на 40-й нед. Масса при рождении 3850 г, рост 53 см, оценка по APGAR 8/9 баллов. Состояние после рождения удовлетворительное. Раннее психомоторное и физическое развитие — по возрасту. Наследственность по нарушению липидного обмена не отягощена.
С декабря 2013 по июль 2015 г. многократно обследован по месту жительства, т.к. отмечались стойкие изменения в виде цитолиза (АЛТ 46,9–154 Ед/л, АСТ 53,3–89 Ед/л), холестаза (общий билирубин 36,6 мкмоль/л, прямой билирубин 5,15 мкмоль/л), нарушения липидного профиля (гиперхолестеринемия (8,4– 9,5 ммоль/л), повышение ЛПНП (7,0 ммоль/л), снижение ЛПВП (1,42 ммоль/л), а также гепатомегалия и нормальные размеры селезенки по данным УЗИ. С 2014 г. (с возраста 5 лет) отмечено увеличение живота в объеме.
Скрининг на гепатиты В и С отрицательный, показатели церулоплазмимина и альфа-1-антитрипсина в норме; маркеры аутоиммунных гепатитов, целиакии отрицательные.
Наблюдался с диагнозом: «Гиперхолестеринемия 2 Б типа. Гепатомегалия неясного генеза». Проведен курс ферментной терапии препаратами панкреатина.
В октябре 2015 г. была заподозрена болезнь Вольмана, рекомендовано проведение энзимодиагностики. Назначена витаминотерапия, пробиотики, желчегон-
125
Атлас редких болезней
|
|
ные препараты. Ферментный анализ |
||
|
|
не сдан. |
|
|
|
|
В июле 2015 г. обследован в НЦЗД. |
||
|
|
При поступлении: масса тела 25 кг, |
||
|
|
рост стоя 126 см. Состояние пита- |
||
|
|
ния удовлетворительное. |
Кожные |
|
|
|
покровы смуглые, чистые от сыпи; |
||
|
|
следы перенесенной |
ветряной |
|
|
|
оспы на передней брюшной стен- |
||
А |
Б |
ке. Слизистые оболочки иктерич- |
||
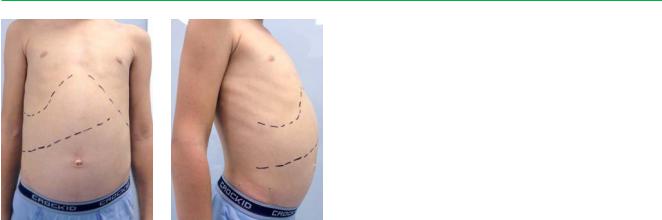
А, Б. Внешний вид ребенка с болезнью накопления |
ные, блестящие. Органы пищеваре- |
|||
ния: аппетит сохранен; язык обложен |
||||
эфиров холестерина: умеренное увеличение живота |
||||
в объеме за счет гепатомегалии |
беловатым налетом; живот увеличен |
|||
в размерах (в объеме 59 см), печень +1+5+8+9+7 см от края реберной дуги, край ровный, эластичный, безболезненный; селезенка не пальпируется (см. рис.).
Результаты обследования
Клинический анализ крови: без патологии.
Биохимический анализ крови: повышение КФК до 1561 Ед/л, АСТ 74 Ед/л, АЛТ 71 Ед/л, гиперхолестеринемия 8,98 ммоль/л, гипертриглицеридемия 2,42 ммоль/л. Уровень лактата крови натощак 3,8 ммоль/л (при норме до 2,2 ммоль/л).
Тандемная масс-спектрометрия (спектр ацилкарнитинов, аминокислот крови) — без патологии.
Исключены альфа-1-антитрипсиновая недостаточность (уровень альфа-1- антитрипсина в норме), Болезнь Вильсона (уровень церулоплазмина 40,2 мг/дл), мукополисахаридоз (анализ мочи на ГАГ в норме), муковисцидоз (потовый тест отрицательный, панкреатическая эластаза в норме), диагноз гликогеновой болезни маловероятен (при контрольных обследованиях уровень гликемии в норме).
УЗИ органов брюшной полости:
•печень: размер выраженно увеличен (+7,5 см из-под реберной дуги), ЛД 76 мм,
первый сегмент 22 мм, ПД 130 мм, контур ровный, край закруглен, угол ~70–75°; паренхима диффузно неоднородная, повышенной эхогенности, поглощение УЗ-сигнала на 1/2; воротная вена расширена до 9,1 мм, кровоток ламинарный 9–10 см/с; в печеночных венах умеренная вертикализация;
•селезенка: измененная; размер увеличен — 107 48 мм; паренхима однородная, средней эхогенности; по ходу корня брыжейки визуализируются немногочисленные лимфатические узлы умеренно пониженной эхогенности максимальным размером до 10 8 мм.
Фиброэластография печени при проведении серии измерений в правой доле
печени: медиана 5,9 кПа; не повышена. Колебания показателей эластичности в отдельных измерениях от 5,3 до 8,9 кПа; IQR 1,3; S. rate 100%. Данных за фиброз печени не получено, F0 по шкале METAVIR.
Энзимодиагностика: активность кислой липазы в лейкоцитах снижена до 14 нМ/мг в час (норма 30–118).
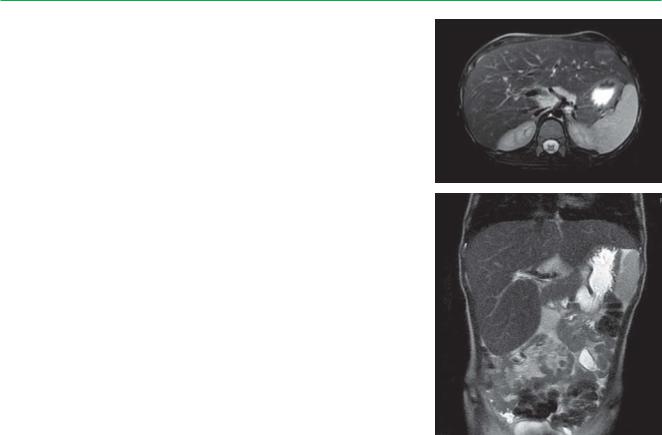
МРТ брюшной полости с МРХПГ: Гепатомегалия. Признаки диффузных и перипортальных изменений печени, склерозирование желчных протоков, реактивные
126
Дефицит лизосомной кислой липазы
изменения поджелудочной железы. Увеличение размеров желчного пузыря, вероятно, как проявление ДЖВП. Лимфаденопатия (см. рис.).
Молекулярно-генетическое исследование. Методом прямого автоматического секвенирования были исследованы все кодирующие экзоны (2–10) гена LIPA, а также прилегающие интронные области. В экзоне 04 выявлена мутация с. 398С>А
вгомозиготном состоянии, приводящая к преждевременной терминации трансляции аминокислотной последовательности p.Ser133X. Мутация ранее не описана, по данным компьютерного анализа (Alamut Visual), является патогенной. Заключение. У пациента выявлена мутация с. 398С>А в гене LIPA
вгомозиготном состоянии, которая может приводить к развитию болезни Вольмана.
Таким образом, данные клинической картины (наличие гепатомегалии), характерные лабораторные изменения (повышение уровня холестерина, триглицеридов, липопротеидов высокой плотности, печеночных аминотрансфераз; снижение уровня липопротеидов высокой плотности), результаты энзимодиагностики и молеку- лярно-генетического исследования позволили установить ребенку диагноз «Дефицит лизосомной кислой липазы. Болезнь накопления эфиров холестерина». Пациент в настоящее время наблюдается в Центре, ему назначена специализированная диета и поддерживающая гепатопротекторная терапия с возможной ферментозаместительной терапией в последующем.
Список рекомендованной литературы
1.Краснопольская К.Д. Наследственные болезни обмена веществ. Справочное пособие для врачей. М.: Центр социальной адаптации и реабилитации детей «Фохат». 2005.
А
Б
А, Б. МРТ органов брюшной полости с МРХПГ у пациента
ввозрасте 6 лет с болезнью накопления эфиров холестерина: печень расположена обычно,
вразмерах значительно увеличена за счет правой и левой доли (занимает передние отделы поддиафрагмального пространства слева) с диспропорцией
II–III сегментов; контуры четкие, ровные; определяются диффузные изменения паренхимы и слабовыраженное перипортальное повышение МР-сигнала за счет соединительнотканно-фиброзных изменений; нижний край печени закруглен; селезенка однородного МР-сигнала, в размерах увеличена, контуры четкие, ровные
2.Hoffman E.P., Barr M.L., Giovanni M.A., Murray M.F. Lysosomal Acid Lipase Deficiency. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle. 1993–2015.
3.Porto A.F. Lysosomal acid lipase deficiency: diagnosis and treatment of Wolman and Cholesteryl Ester Storage Diseases. Pediatr Endocrinol Rev. 2014 Sep; 12 (Suppl. 1):125–32.
4.Bernstein D. L., Hulkova H., Bialer M. G., Desnick R. J. Cholesteryl ester storage disease: review of the findings in 135 reported patients with an under diagnosed disease. J Hepatol. 2013 Jun; 58 (6): 1230–43.
127
ДИСПЛАЗИЯ ФРОНТОНАЗАЛЬНАЯ
(FRONTONASAL DYSPLASIA)
МКБ-10: Q75.1; ОМIМ 304110, 305645
Определение. Дисплазия фронтоназальная — порок развития из группы череп- но-лицевых дисплазий с обширной группой дефектов развития лицевого скелета.
Впервые фронтоназальную дисплазию описал Hoope в 1859 году, а спустя век (1967) W. DeMeyer определил патологический процесс как «синдром срединной расщелины лица». Учитывая частое сочетание фронтоназальной дисплазии с дефектами костей свода черепа и аномалиями головного мозга, М. Cohen в 1979 г. предложил общее название этих случаев — краниофронтоназальная дисплазия.
Синонимы: срединная расщелина лица, дисплазия краниофронтальная, дизостоз краниофронтоназальный, синдром краниофронтоназальный.
Эпидемиология. Распространенность в популяции составляет 1:70 000– 100000 населения.
Тип наследования: неизвестен. Большинство случаев — спорадические. Описаны случаи с аутосомно-доминантным, Х-сцепленный доминантным типом наследования.
Этиология, патогенез. В основе таких пороков лежит нарушение развития вентральных отделов 1-й жаберной дуги.
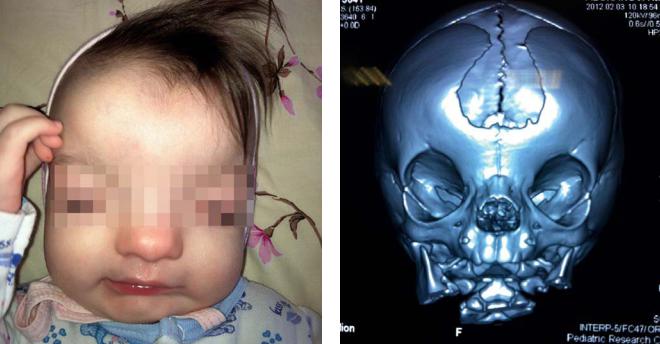
Внешний вид больной с фронтоназальной |
Компьютерная томография костей черепа больной |
дисплазией |
с фронтоназальной дисплазией |
128

Дисплазия фронтоназальная
Клинические проявления
Синостоз венечного шва, брахицефалия, гипертелоризм, антимонголоидный разрез глаз, расщепление кончика носа, широкая спинка носа, высокое арковидное небо, расщелина губы/неба, удлиненные вдавленные ногти, жесткие волосы, аномалии пальцев кистей и стоп (широкий I палец, синдактилия), псевдоартрозы ключиц, «куриная грудь», диафрагмальная грыжа, шалевидная мошонка, гипоспадия. Иногда наблюдается полное удвоение носа.
Синдром срединной расщелины лица — один из 4 синдромов, при которых регистрируется эндокринная патология: задержка роста (церебральный гипофизарный нанизм); вторичный гипотиреоз; двусторонний крипторхизм; патология полового члена (сигарообразная форма); вторично-первичный гипогонадизм; уровень лютеинизирующего гормона в пределах 1 МЕ/мл, фолликулостимулирующего гормона — до 1 МЕ/мл; результат пробы с люлиберином — отрицательный; проба с хорионическим гонадотропином показывает повышение содержания тестостерона, но ниже возрастных норм; азооспермия.
Костная система: отсутствие половины позвонка, spina bifida.
Орган зрения: колобома, врожденная катаракта, эпибульбарные дермоиды и липомы, микрофтальмия и анофтальмия.
Орган слуха: деформация, гипоплазия или аплазия наружного уха, периаурикулярные кожные выросты, низко расположенные ушные раковины, проводящая глухота.
Центральная нервная система: гидроцефалия, микрогирия, лиссэнцефалия, агенезия мозолистого тела, краниостеноз, приводящие к различной степени выраженности умственной отсталости.
Диагностика
•Клинические проявления;
•ранняя УЗ-диагностика патологии лица у нерожденного ребенка;
•результаты рентгенологического обследования;
•кариотипирование (консультация генетика);
•компьютерная томография (по показаниям).
Лечение хирургическое (при возможности).
Прогноз различен: для фронтоназальной дисплазии 3-й степени неблагоприятный, дефекты 1 и 2-й степени подлежат хирургической коррекции.
Список рекомендованной литературы
1.Adolphs N., Arnaud E., Haberl E.J., Graul-Neumann L., Schmidt G., Menneking H., Hoffmeister B. The oculo-auriculo-fronto-nasal syndrome (OAFNS) — Description of a rare and complex craniofacial deformity and its interdisciplinary management before school age. Jornal of сranio–maxillo–facial surgery. 2012, Dec; 40 (8): 668–74.
2.Tonni G., De Felice C., Asioli S., Ventura A. Fronto-nasal dysplasia and atrio-ventricular canal in a fetus with trisomy 18 identified by absent nasal bones during first trimester screening scan. Congenital anomalies. 2007 Mar; 47 (1): 45–8.
3.Suthers G., David D., Clark B. Fronto-facio-nasal dysplasia. Clinical Dysmorphology. 1997, Jul; 6 (3): 245–9.
129