

3 курс / Патологическая физиология / Основы общей патологии
.pdf! |
441 |
восстановлеповрежденныхсосудовика тромбаиеализацию. Взаимодейсттромбом,лейкоцитыразвязокругуянеговают процевоспале.(нижем)Наконец. нельзя, ияотметить,что большинплазменныхфакторвёртыванияство, оречьторых пойдётним, жечастичноютмакрофагальноепроисхождение.
Н. И. Цыбиков(1986)придаетбольшоезначениеест ственным аутоантителампро активныхформферментовсистемы фибрина,спомкоторыхщьюлимфоцитымогутпринимать участиевпроцессахгемостаза.
|
Принедостаклемеханизмовточнгемостазаыхости |
|
|
развиваетсягеморрагичессиндр,котха ыйктеризуетсяий |
|
||
нарушениемфункций( |
тромбоцитопатия)и/илипонижением |
|
|
количестватромбоцитов( |
тромбоцитопения)Приэтом. ,как |
|
|
правило,отмечаетсякровоточивостькапиллярноготипасм(.выше), |
|
|
|
кровоизлформнемедлипослеяниеруетсяповреждениянно |
баиретракция |
|
|
микрососудов,нарушобрбазованиетромлоготся |
|
||
кровяногосгустка.Эндотелипробыположи,атесльн, тыельны |
|
|
|
отражающие [255] работусистемыфибрина,изменены. |
- |
||
Основныепроявлениясм.( |
таблицу 9)включаютпетехиально |
||
экхимозную сыпьнакожеислизи,приболеесерьёзнойтых |
|
||
недостаклезвточногемостазанабытьстиут |
|
|
|
кровотеченияслизистыхэпистаксис(,мелена,гемоптоэ, |
-затого,чтожизненный |
|
|
метроррагия)Гематомы. нехарактерны.Из |
|
||
циклтромбоцитовкороток,какправ |
ило,адекватлечениепри ое |
|
|
этойфоргеморрагичесиндромадавебытсьмакоготрый |
|
|
|
эффект. |
|
|
|
|
Тромбоциможевозпонтикатьесколькименияпричинам. |
|
|
Увеличенселезёнкиактивностиеёмакрофагов,известноекак |
ластинокв |
|
|
гиперспленизм,ведеткнакоплениюкровяных |
|
||
селезёночныхсосудахитромбоцвперикровфертопениической |
|
||
(см.. |
224)Улавливаютсятромбоциты. поверхностью |
|
|
злокачественныхопух,напримерлейпухолиВильмса.Д угими |
|
|
|
возможнымипричинамитромбоцитопенииявляютсяугнетение |
|
|
|
мегакариопоэза (апластическиемегалобластическиесостояния, |
|
||
инфильтрациякостногомозгалейкозныбластаопухолевыми клетками,миелоф,изолугнетеиброзровамегакариопоэза,ниеное например,уалкоголикеилиприёмефенотиазидных
! |
442 |
диуретиков), а также ускоренная гибель тромбоцитов (потребление при распространенном и системном тромбогенезе или септических процессах, аутоиммунный цитолиз и аутофагоцитоз).
Если содержание кровяных пластинок снижено не сильно (не ниже 90-100 тыс. в мкл) — тромбоцитемия может протекать бессимптомно и проявляться только удлинением времени кровотечения при пробе Дьюка. При количестве тромбоцитов от 100 до 50 тыс. на мкл. имеется постоянная пурпура и возможны нетяжёлые кровотечения из слизистых, например, повторяющиеся носовые. О. Франк считал критическим числом тромбоцитов 35 тыс. на мкл (число Франка). Критический уровень тромбоцитов определяется сопутствующими заболеваниями а другими условиями индивидуально. Во всяком случае, при тяжелых тромбоцитопениях, ниже 20 тыс. в мкл имеются опасные проявления кровоточивости: спонтанные геморрагии из слизистых, постоянная кровоточивость дёсен, гематурия и могут даже развиваться внутричерепные кровоизлияния. Если число тромбоцитов менее 5 тыс. в мкл, то тяжелая картина тромбоцитопении дополняется и некоторым понижением скорости фибринообразования.
Наиболее частая причина тромбоцитопений это аутоиммунные процессы, поражающие кровяные пластинки. Выше уже упоминалось о болезни Верльгофа — хронической иммунопатологической тромбоцитопенической пурпуре (ХИТП). Этиология аутоиммунного процесса при данном заболевании не установлена.
Критериями болезни являются аутоантитела к тромбоцитарному гликопротеину gpIIIa, поражающие тромбоциты и мегакариоциты, мегакариоцитоз в костном мозге, увеличение размеров кровяных пластинок и явления гиперспленизма. Симптоматическая ХИТП может сопровождать аутоиммунные заболевания с неорганоспецифическими аутоантителами (системная красная волчанка).
Острая аутоиммунная тромбоцитопения может быть спровоцирована иммунокомплексным процессом с участием нетромбоцитарных антигенов, когда иммунные комплексы
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
443 |
осаждаютсянатромбоцирецепантителускоряюторахных аутофагоциткровяпластинок.Векыхслучаязторых, взаимодействиетромбоцитагаптена,либоэкзогенногонтигена ведёт кпоявлениюнеоантигенов,провоцирующихиммунный тромбоцитолиз.Этимеханизмыобуславливаютострую тромбоцитопеническуюпурпуруприв русныхинфекциях (краснуха,мононуклеоз,гепацитомегалов, инфекц), ирусная
приёмерядалекарствхинин(,хинидин, сульфаниламиды, аллопуринол,парацетамол,мети,рифампицДОФА,дигокс, ин бензодиазепины,гепариндр.)Вэтом. случаетромбоцит — невиннаяжертвасвоеголюбопытства« »,выражающегося уникальноширокнабореповерхностныхмрецепторов.
Особеннобольшоез |
начениеимеетсиндромг паринзависимой |
[256] |
||
тромбтромбоцитопеническойтическойпурпуры |
.Ондавно |
|||
известенрачамипреждерасц,какпарадоксальнаянивался |
|
|
||
реакциянагепарин.Гепаринвзаимодействуеттромбоцитарным |
оиммуннымпроцессам |
|
||
факторомУ4лиц.снаклонностьюаут |
|
|
||
комплексгепаринафактора4поверовяныхплахностинок |
|
|
|
|
становится,какнеоантиген,объекиммуннойсоомакистороны |
|
|
— |
|
IgG,котмоарыегрегироватьутромбоциты,таккакихFab |
|
|
||
фрагментыфиксируютпластиповерхностнымки |
|
оантигеном, |
||
Fc-фрагментсоединяетсятромбоцитарнымиммуноглобулиновым |
|
|
||
рецептордругтр .Омгобмразуютсяоцитаартериальные |
|
|
|
|
конглтром,вызывающиеератыбоцм кротромбоэмболиютов. |
|
|
||
Из-запотребкровялаеихнсодеыхтинокиявржаниеови |
|
|
|
|
снижается. |
Вследствактивациитромбоцитовиммуннымие |
|
|
|
комплексзамыкпорочныйкругаипроцессмиетсяделаетсявсё болеераспространённым.
Сходнаякараспросттинартеранениального тромбоитромбоцитопениябразованияпотренаблюдается
приидиопатическойтр омботическойтромбоцитопенической пурпуре,длякоторойпровоцирующийагентнеизвестен.Больные
имеютаномальныйкомплексvWF -VIII-Cицитотоксические аутоантителакэндотелиоцитам,вызывающиер спространенную агглюттромбоциихфинациюкарсациютовери альными капиллярныммикропочекзгасосудам.Этовызывает внутрисгем,посудистыйявлензг алиновыхсгусткове
! |
444 |
тромбов, содержащих кровяные пластинки и фибрин, неврологические и почечные нарушения, вторичную активацию фибринолиза. Форма с преобладанием поражения сосудов почечных клубочков описывается как гемолитико-уремический синдром.
Эти нарушения имеют могут интерпретироваться, как региональные формы ДВС-синдрома.
Наконец, иммунопатологическая тромбоцитопения может возникать при изоиммунных конфликтах. У матерей, не имеющих тромбоцитарного антигена PLA1, плод, располагающий этим антигеном отцовского происхождения, подвергается риску
тромбоцитолитической болезни новорожденных, аналогичной широко известному синдрому изоиммунной гемолитической анемии, вызванной резус-конфликтом матери и плода. Такая же ситуация может иметь место при переливании крови, несовместимой по тромбоцитарным антигенам и у новорожденных от матерей с болезнью Верльгофа.
Если присутствуют проявления кровоточивости по капиллярному типу, а количество тромбоцитов нормально, то речь может идти о функциональной неполноценности кровяных пластинок — тромбоцитопатии.
Тромбоцитопатии могут быть наследственными и приобретёнными.
Функции тромбоцитов нарушаются под действием лекарств (салицилаты, дериваты фенилбутазона), при уремии, при гемобластозах, при болезнях печени, у алкоголиков.
Нестероидные противовоспалительные агенты нарушают синтез простагландинов и тромбоксана А2, что вызывает пострецепторный блок многих внутритромбоцитарных механизмов активации и тормозит адгезию, агрегацию и реакцию освобождения, а также вязкий метаморфоз. Это единственная ситуация, когда in vitro тромбоциты не склеиваются под воздействием арахидоновой кислоты. Уремические токсины, в частности, аргининянтарная кислота и феноловые производные, нарушают экспонирование тромбоцитарного 3 фактора и ухудшают сродство vWF к тромбоцитарным рецепторам. Тормозятся и адгезия, и агрегация.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
445 |
Пригемобластозмогутнаблюдатьсяблокирующиеаутоантителах |
ам,например,крецепторамфактора |
ктромбоцирецептаорным |
|
vWFприл мфопрзабол.Другойиферативныхпричеваняхной |
|
тромбоцитопаприлейкозахявляютсяаномалиисозреванияий |
|
гранулвмегакариоцитах.Приалкоголизмесильновозрастает |
пластывание |
вязкостьклеточныхмембран,чтозамедляетрас |
|
тромбиреакциюосвобождецитов.Вероят,какиенияо |
-то |
токсичдейсиепродуиприкпечёночнойтвуюты |
|
недостаточ.Показано,чтоприхроническомгепатитести тромбоцитыдегранулируютсявпечени.Прихол тергстазентное
[257] действиежелчны |
хкислотнеблагоприятносказываетсяна |
функцияхтромбоцитмембраны. рной |
|
Еслипр обретённыетромбоцитопатииком, лриксны |
|
наследственныхчащенаблюдаютсяизолированные |
|
функциональдефе.Каужеговорилосьтывыш,нарушенияые |
вклетчастоказываютсяепричиной |
инфопрмационныхоцессов |
|
еенеспособностиполноправильносуществить |
|
запрограммировответ.Иллюстрэтоготезисамогутнныйцией |
|
послужитьтромбоцитопатии,прикоторыхотдельные |
-за |
тромбоцитарныефункциимогутвыпадмоз,изичноть |
|
невозможносуществитьтирасп илизнаваниеиногоси.гонала |
|
Втакойситуациибольшоезначениеприобретает |
|
диффередиагспомощнциалостикатестированияьнаяю |
таблица 8). |
отдельныхтромбоцитарныхфункцийin vitro ( |
|
Наслдедствфектытромбоцитовделятсянпатологиюные адгезии,агрегосвобожденияакцции.
Наиболееважнаяраспространённаявпрактикенаследственная
адгезиотромбоцитопанаблюдаетсянаяпри ия |
|
болезнифон |
Виллебранда,частотакоторойд |
оходитдо1/800Даннаяомалия. |
|
имеетфо3,прнаиболеемычастойизкоторых,аутосомно |
|
- |
доминантной,патологиязаключается,какужеговорилосьвыше, |
|
|
недостаточнойэкспрессииvWF,чтопозволяеттромбоцитам |
к vWFявляется |
|
эффективнопри эндотелиюлеив.Такться |
|
|
носителемфактораVIII |
-Cизащищаетегопротеолиза,убольных |
|
частоимеетсянетольснижениеконцентрацииvWF,но |
|
-C,поэтомуихрасстргемойствостаза |
некотораянехваткаVIII |
|
|
! |
446 |
приобретает черты комплексного и сочетает дефект адгезии тромбоцитов и пониженное частичное тромбопластиновое время, что относится уже к коагулопатии. Другой, редкий вариант болезни вызван нарушением полимеризации мономерных молекул vWF. Уровень vWF и VIII-C нормален, но скорость связывания тромбоцитов с vWF снижена. Адгезия уменьшена, но нарушения коагуляции нет. Еще реже встречается третий, наиболее тяжёлый вариант — двойная гетерозиготность по паре разных рецессивных мутаций vWF или же гомозиготный рецессивный вариант болезни. Пациенты этой группы не имеют vWF и, вдобавок, страдают выраженным [258] гемофилиоподобным нарушением коагуляции, так как VIII-C фактор у них является очень короткоживущим.
Дефект гликопротеина gp Ib-Ix проявляется аутосомно- рецессивным синдромом Бернара-Сулье. Нарушается адгезия, срок жизни тромбоцитов укорачивается, а их средний размер сильно возрастает. Следовательно, тромбоцитопатия сочетается с тромбоцитопенией. Сочетание количественной нехватки и качественного дефекта тромбоцитарной реакции освобождения характерно и для синдрома Вискотта-Олдрича, при котором нарушения образования белого тромба сочетаются с иммунодефицитом (см. ниже «Иммунодефицитные состояния»)
Дефект гликопротеина gp IIb-IIIa ведет к тромбастении Гланцманна. Тромбоциты больных нормальны на вид, способны адгезировать, освобождать медиаторы и распластываться на коллагене, но теряют возможность связывания фибриногена и плохо агрегируют. Отсутствие фиксации к фибриногену не дает и возможности для полноценной ретракции, несмотря на нормальное количество кровяных пластинок.
При кожно-глазной форме наследственного альбинизма,
синдромах Германского-Пудляка и Чедиака-Хигаши (см. ниже,
раздел «Фагоцитоз») нарушается реакция освобождения гранул тромбоцитов, так как имеется дефицит структурного белка тромбоцитарных б-гранул либо компонентов плотных телец, с их морфологическими аномалиями.
Из-за нарушения дегрануляции ослабляется агрегация, особенно,
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
447 |
обеспеченнаяиндуцируемымирецепторамиколлагенаАДФ,но адгезияответнаристомицинарахк остаютсядоновуюслоту норме.Наруше нияформигранулпэтсованияндромах распространяютсяинагранулоциты.
|
Притестированиифункцийтромбоциследуеучитывать, ов |
|
|
|
|
|
|||
чтоАДФ,норадреналин,тромбин,коллаген, , алин |
|
|
|
|
|
||||
арахкиидоноваяслорисагглютином хinцинПрvitroируют. |
|
|
|
|
-IIIa,а |
||||
этом,ристомииспользуетрецепторадгезииинЙЙвgp |
|
|
|
|
|||||
остальныедействуютчерезфибриногеновыйецептор,что |
|
|
|
|
|
||||
позволяетпроводдифференцировкутьзолированных |
|
табл. |
8). |
||||||
функциональныхдефектовкровяныхпластинок( |
|
||||||||
|
Тромбоц,атакжеакттромбоцитовтозвациялейкоцитов |
|
|
|
|
|
|||
крподвоздействвитромб,липополисахаридовнабем,ктерий |
|
|
— |
|
приводятк |
||||
другихполианмоле,какгемолизонныхул |
|
|
|
|
|||||
эксцессамвработеклеточнзвгемостазанаиспособствуютго |
- |
исгусткообразованию. |
|||||||
распространёнисистромбоемному |
|||||||||
Причинытромбсуждалисьоцитозавыше.Отметимповышенное |
|
|
|
|
|
||||
тромбприостромобразмиелейкозМбластномвание7 |
|
|
|
|
|
||||
(мегакариобластныйлейкоз)МЗпроми( )Вйкоз.лоцитарный |
ируютпромиелоцклетки, |
тарные |
|
|
|||||
послпролифереднемучае |
|
|
|
||||||
исключительнобогатыепрокоагулянтнымифосфолипидами |
|
|
|
|
|
||||
(«кровянымтромбопластинТромбоцитоз»). сопров ждает |
|
|
|
|
|
||||
некоторыевариахронмтыическогоелолейкоза.Избыточная |
|
|
|
|
ви |
||||
работагемостатическихмехприактивациинизмовклеткро |
|
|
|
|
|||||
эндотелияцитокинамирассматриваетсяследующразделепри |
|
|
|
|
|
||||
обсужденииДВС |
-синд.Передрассмотомафибриениемового |
|
|
|
|
||||
гемостазаикоагулопатий,приводосновныекритериим |
|
|
|
|
|
||||
диффередиагеморрагическогонциальнойостндромаки |
|
|
|
|
|
||||
(табл. |
9). |
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
НАРУШЕНИЯГЕМОСТАТИЧЕСКИХ АНТИГЕМОСТАТИЧЕСКИХМЕХАНИЗМОВ
ПЛАЗМЫ
Плазмакрсовиднеактивныержитпредшественники |
|
протеолитическихферментов,основном,относящиесяфракций |
онтактесполианионными |
в-глобулинов,которыеспособныпри |
! |
448 |
поверхностями и молекулами, а также под влиянием других протеаз, входящих в сторожевую полисистему плазмы крови (см. раздел «Патофизиология воспаления»), переходить в активное состояние путём ограниченного ступенчатого протеолиза, последовательно самособираться в комплексы и, в итоге, обеспечивать формирование стабильного полимерного фибрина, армирующего тромб. [259]
Эта часть процесса тромбообразования называется свёртыванием крови (гемокоагуляцией), и для его осуществления на всех этапах, кроме первых двух реакций внутреннего каскада, необходим ионизированный кальций. Это позволяет предупреждать фибринообразование in vitro с помощью таких агентов, как цитрат, оксалат и ЭДТА. Помимо кальция каждый шаг в ступенчатом механизме свёртывания предусматривает участие проактиватора, протеазы, ее кофактора и фосфолипидного ложа, предоставленного для реакции сосудистой стенкой, тканями и тромбоцитарно- лейкоцитарным тромбом. В табл. 10 представлены краткие сведения о плазменных факторах свёртывания.
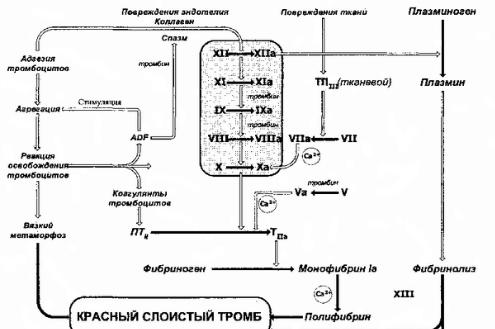
Образование фибрина происходит в три этапа Вначале образуется мембранносвязанный комплекс протеаз и белковых кофакторов, обладающий свойствами протромбиназы (рис. 61). Нарушения этого этапа носят название тромбопластинопатии.
Рис. 61. Механизмы фибринообразования и способы его контроля.
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/
! |
449 |
Затемпротркомпревращаетплексбиназныйбелок |
|
|
|
|
|
|
|
- |
|||
предшественнпротромбин.Дефеэтогоэтапкты |
|
|
|
|
Тромбин,всвоюочередь,действуя |
а |
|||||
свёртывания — тромбинопатии. |
-мономер. |
||||||||||
какферментнафиб,п иногеневращаетегофибрин |
|
|
|
|
|
|
|
|
|||
Полимеризациямономеравфибрин |
|
|
|
|
|
-полимеробразование |
|||||
фибрин-полимереперекрёстныхсшивокподдействиемXIII |
|
|
|
|
|
|
|||||
фактораведутформ |
|
|
|
ированиюфибриновойчаститромба. |
|
|
|
|
|||
Патологиязавершающегоэтапафибриноотнк ситсябразования |
|
|
|
|
|
|
|||||
фибринопатиям. |
|
|
|
|
|
|
|
|
|
||
Образованиепротркомплексабиназного |
|
|
|
|
|
— наибсложныйлее |
|||||
идлительныйэтапфибриногенеза. |
|
|
|
|
|
|
|
|
|
||
Оносуществляетсякакответнаповреждениетка( й |
|
|
|
|
|
|
|
шний |
|||
каскад),такивответнаконтакткомпонентовплазмыс |
|
|
|
|
|
|
|
|
|
||
полианионамивнутренн( каскад). й |
|
|
|
|
|
|
|
|
|
|
|
Реакциивнешнегокаскадаболеебыстроприводятк |
|
|
|
|
|
|
|
|
|
||
формирпротромби, собенван,привыраженномю азыо |
|
|
|
|
|
|
|
е |
|||
повреждениитканей.Приразмозжённыхранахфибринообразовани |
|
|
|
|
|
||||||
можетзакончитьсявпределахминуты1.Внутреннийкаскад |
|
|
|
[260] |
Рис. |
61. |
Механизмы |
||||
осуществляетсяболее |
|
|
|
|
|||||||
фибриспоснегкобразнтролябывания |
|
|
|
|
|
|
. |
[261] медленно,за |
|||
2-6мин.нопри, м нимальныхтравм |
|
|
|
|
|
ахсосудов,например, |
|||||
хирургичесразрезах,эффефибрикихтивбезнообразование |
|
|
|
|
|
|
|||||
негоневозможно.Отчасти,именнопоэтомудляпациентовс |
|
|
|
|
|
|
|
|
|
||
гемофилиейАВуш размозжённыебленныеранымогут |
|
|
|
|
|
|
|
|
|
||
представлятьменьшуюопасносмыпрофузныхстьле |
|
|
|
|
|
|
|
|
|
||
кровотечений, |
чемпростыепорезы.Приактивациивнешнего |
|
|
|
|
||||||
каскадарольмембраннойподложкииграетсосудистая,енкапри |
|
|
|
|
— |
фосфолипидымембран |
|||||
реакцияхвнутреннегокаскада |
|
|
|
|
|
||||||
формэлементовкровинных. |
|
|
|
|
|
|
|
|
|
|
|
Обакасхоканаключевомдятсяаэтапе |
|
|
|
|
|
|
|
|
— активацииX |
||
фактора. |
|
|
|
|
|
|
|
|
|
|
|
Какпока |
занона |
рис. |
61, |
вовнешнемкаскаде |
|
|
VIIфактор |
||||
|
|
|
|
|
|
|
|
|
|
|
|
прискотредзаряженнымицательняетфосфолипидам
! |
450 |
тканевого тромбопластина за счёт хальций-фиксирующего участка, содержащего остатки уникальной ди-γ-карбоксилглютаминовой кислоты. Это ведет к активации VII фактора. VIIa фактор в дальнейшем контактирует с X и IX фактором и активирует их. IХа фактор способствует дальнейшей активации X, формируется фактор Ха.
При работе внутреннего каскада свёртывания на поверхности повреждённого сосуда, содержащей коллаген, самособирается комплекс из трёх белков сторожевой полисистемы плазмы крови; высокомолекулярного кининогена, прекалликреина и XII фактора. При этом XII фактор превращается в XIIa, действуя как фермент на прекалликреин и XI фактор, преобразуя их в калликреин и ХIа. Калликреин усиливает процесс, подвергая протеолитической активации дополнительные порции XII. Одновременно, концевой пептид, потерянный фактором Хагемана, запускает процесс фибринолиза. ХIа протеолитически активирует фактор IX, который в присутствии антигемофильного глобулина VIIIa-С переводит X в Ха. Фосфолипидную подложку могут создавать клетки формирующегося белого тромба.
Наконец, активный Ха расщепляет на поверхности тромбоцитов (в присутствии их 3 фактора и Va-кофактора плазмы) протромбин с образованием тромбина.
Тромбин оказывает мощное обратное усиливающее влияние на активацию тромбоцитов, агрегацию, частичный протеолиз V, VIII и X факторов, но основной его функцией является преобразование I фактора в фибрин. Он также активирует XIII фактор, трансглютаминазу, сшивающую фибриновые нити, и запускает ретракцию. Тромбин — медиатор воспаления (вазоконстриктор, хемоаттрактант и активатор лейкоцитов).
Система внутреннего каскада может быть запущена лизосомальными протеазами и протеолитическими факторами комплемента при воспалении и аллергических реакциях. Это показано на рисунке 62, отражающем интегральную схему гемостаза.
Наследственные и приобретенные нехватки факторов
Рекомендовано к покупке и изучению сайтом МедУнивер - https://meduniver.com/