

Аналитика. Электрохимические методы анализа
.pdf2.4. ПРЯМАЯ ВОЛЬТАМПЕРОМЕТРИЯ
Прямая вольтамперометрия предполагает использование зависимостей потенциал – ток, полученных в электролитической ячейке с особым электродом, кроме ртутного капающего. Принципы выполнения качественного и количественного анализа те же, что и в описанном ранее полярографическом методе.
Индикаторным обычно служит вращающийся платиновый электрод или электрод из графита и графитовых материалов (стеклоуглеродный, углеситалловый).
Эти электроды отличаются от капающего ртутного тем, что они имеют другую область поляризации и поверхность их в процессе получения вольтамперограммы не возобновляется.
На рис. 20 приведены вольтамперные характеристики платинового, графитового, ртутного электродов.
Известно, что рабочая область потенциалов электродов ограничивается потенциалами разряда фонового электролита.
Рис. 20. Вольтамперные кривые для платинового, графитового и ртутного индикаторных электродов (Iк – катодный ток, Iа – анодный ток)
41

Ртутный электрод благодаря высокому перенапряжению выделения водорода можно использовать в области больших отрицательных потенциалов (табл. 6). На графите и платине рабочая область ограничена более низкими отрицательными потенциалами. Указанные рабочие области приведены в табл. 7, 8.
|
|
|
|
|
|
|
|
|
|
|
|
|
Таблица |
6 |
|
|
Рабочая область потенциалов ртутного электрода |
|
|
||||||||||||||
|
|
|
|
(относительно н.в.э.) |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Фон, 1 н. раствор |
|
|
Потенциалы, В |
|
|
Фон, 1 н. раствор |
|
Потенциалы, В |
|
|
||||||
Н3РО4 |
|
|
+0,6 –1,1 |
|
|
|
K2SО4 |
|
+0,6 –1,5 |
|
|
|||||
H2SO4 |
|
|
|
|
|
|
|
KNО3 |
|
|
|
|
|
|||
|
|
+0,6 –1,1 |
|
|
|
|
+0,8 –1,5 |
|
|
|||||||
HNO3 |
|
|
+0,8 –1,1 |
|
|
|
KСlО4 |
|
+0,8 –1,5 |
|
|
|||||
НСlО4 |
|
|
+0,8 –1,1 |
|
|
|
KСl |
|
+0,3 –1,5 |
|
|
|||||
НСl |
|
|
+0,3 –1,1 |
|
|
|
KОН |
|
+0,1 –1,5 |
|
|
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Таблица |
7 |
|
|
Рабочая область потенциалов платинового электрода |
|
|
||||||||||||||
|
|
|
|
(относительно н.в.э.) |
|
|
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
Фон, 1 н. раствор |
|
|
Потенциалы, В |
|
|
Фон, 1 н. раствор |
Потенциалы, В |
|
|
|||||||
Н3РО4 |
|
|
+1,7 0,0 |
|
|
|
K2SO4 |
+1,3 –0,4 |
|
|
||||||
H2SO4 |
|
|
|
|
|
|
|
KNO3 |
|
|
|
|
||||
|
|
+1,7 0,0 |
|
|
|
+1,3 –0,4 |
|
|
||||||||
HNO3 |
|
|
|
|
|
|
|
KСlO4 |
|
|
|
|
||||
|
|
+1,7 0,0 |
|
|
|
+1,3 –0,4 |
|
|
||||||||
НClO4 |
|
|
|
|
|
|
|
KСl |
|
|
|
|
||||
|
|
+1,7 0,0 |
|
|
|
+1,2 –0,4 |
|
|
||||||||
НСl |
|
|
|
|
|
|
|
KОН |
|
|
|
|
||||
|
|
+1,3 0,0 |
|
|
|
+0,9 –0,8 |
|
|
||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
Таблица |
8 |
|
|
Рабочая область потенциалов различных графитовых |
|
|
||||||||||||||
|
|
|
электродов (относительно н.в.э.) |
|
|
|
|
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
Графит |
|
|
Графит |
|
|
|
|
|
|
|
||
Фон, 1 н. раствор |
|
|
|
|
импрегниро- |
|
Стеклоуглерод |
Углеситалл |
|
|||||||
|
непропитанный |
|
|
|
||||||||||||
|
|
|
|
|
|
|
ванный |
|
|
|
|
|
|
|
||
H2SО4 |
|
|
|
+0,8 –0,1 |
|
+1,4 –0,6 |
|
|
– |
|
|
|||||
|
|
|
|
+1,54 –0,9 |
|
|
||||||||||
HNO3 |
|
|
|
+0,8 –0,1 |
|
+1,4 –0,6 |
|
– |
|
– |
|
|
||||
НСlО4 |
|
|
|
+0,8 –0,1 |
|
+1,4 –0,6 |
|
|
|
+1,34 –0,6 |
|
|||||
|
|
|
|
|
– |
|
|
|||||||||
НСl |
|
|
|
+0,8 –0,1 |
|
+1,1 –0,6 |
|
– |
|
– |
|
|
||||
Na2SО4 |
|
|
|
+0,8 –0,2 |
|
+1,4 –0,8 |
|
+1,44 –0,9 |
– |
|
|
|||||
|
|
|
|
|
|
|
||||||||||
KСl |
|
|
|
+0,8 –0,2 |
|
+1,1 –0,8 |
|
– |
|
– |
|
|
||||
KОН |
|
|
|
+0,6 –0,4 |
|
+0,8 –1,0 |
|
+0,74 –1,1 |
– |
|
|
|||||
42

В области анодных потенциалов применение ртутного электрода ограничено потенциалом электрохимического растворения ртути. Этот процесс довольно легко протекает при потенциале ~0,1 В в щелочной среде и ~0,3–0,6 В в кислой. По этой причине ртутный электрод не применяется в анодной области потенциалов. Как видно из табл. 7, 8, для анодных процессов наиболее пригодны электроды из платины и графита.
Поверхность твердых индикаторных электродов часто загрязняется продуктами электродных реакций. Поэтому для получения воспроизводимых результатов необходимо регулярно проводить очистку поверхности электродов. Для этого используют как механические (полировка абразивной пастой), так и химические (обработка кислотой) способы. Для графитового электрода используют также срезание торцевого слоя.
Прямая вольтамперометрия используется в анализе как неорганических, так и органических соединений, содержащих разные функциональные группы: гидроксо-, оксо-, тио-, аминогруппы идругие. Примеры органических соединений, определяемых описываемым методом, приведеныв табл. 9.
Таблица 9
Органические соединения, окисляющиеся на графитовом (платиновом) электроде
Соединение |
Фоновый электролит |
Е1/2, В |
||
Аскорбиновая кислота |
1 М H2SО4 |
0,8 |
|
|
ЭДТА |
0,1 МHCl |
0,7 |
|
|
Фенолыиихпроизводные |
Буферныйраствор, рН= 2–8 |
0,6–0,3 |
||
Аминокислоты |
0,01 МH2SО4 |
1,1 |
|
|
(оксисфенилаланин) |
|
|||
|
|
|
|
|
8-Меркаптохинолин |
Ацетатный буферныйраствор, рН= 4 |
|
0,6 |
|
Тиодиуксусная кислота |
Ацетатныйбуферныйраствор, рН= 4–6 |
|
1,0 |
|
43

2.5. ИНВЕРСИОННАЯ ВОЛЬТАМПЕРОМЕТРИЯ
Принцип инверсионной вольтамперометрии заключается в электрохимическом концентрировании пропорциональной части определяемого вещества на электроде с последующим электрохимическим растворением концентрата. Концентрирование (электролиз) определяемого компонента производят при потенциале, отвечающем предельному току восстановления или окисления вещества. Электролиз ведут в течение фиксированного времени. После концентрирования включают развертку потенциала в направлении более положительных потенциалов по сравнению с потенциалом электролиза. Форма получаемой вольтамперограммы показана на рис. 21.
Рис. 21. Инверсионная вольтамперограмма
Потенциал, отвечающий экстремуму пика, дает качественную характеристику определяемого компонента, а площадь (высота) пика – количественную. Для выполнения количественного анализа аналита используют как метод калибровки, так и метод внутренней нормализации. Метод калибровки заключается в нахождении зависимости площади (высоты) пика от концентрации анализируемого вещества. Калибровку проводят вы-
44
полняя анализ стандартных растворов анализируемого компонента различной концентрации. Затем строят калибровочный график в координатах C–S или C–h (C – концентрация, S – площадь пика, h – высота пика).
Метод внутренней нормализации предусматривает отнесение величины количественного параметра пика (S или h) к сумме указанных величин всех полученных пиков, выраженное в процентах.
Если проводится регистрация вольтамперограммы анодного растворения продукта электролиза, то этот вариант метода называют анодной инверсионной вольтамперометрией.
В методе катодной инверсионной вольтамперометрии анализируемый компонент концентрируют на электроде в виде продукта окисления. Так, марганец можно концентрировать в виде MnO2 при потенциале, отвечающем площадке предельного тока окисления марганца (II) до марганца (IV). Затем производят развертку потенциала в направлении его более отрицательных значений, регистрируя катодную вольтамперограмму восстановления полученного продукта.
Наряду со стационарным ртутным электродом в этом методе применяют графитовые и стеклоуглеродные электроды для концентрирования металлов. Находят применение электроды из графита с нанесенной пленкой ртути (пленочный ртутно-графитовый электрод).
Метод инверсионной вольтамперометрии позволяет определять компоненты пробы при их совместном присутствии. В этом случае концентрирование ведут при потенциале, отвечающем площадке предельного тока наиболее трудно восстанавливающегося вещества. При растворении получается несколько пиков. В качестве примера на рис. 22 приведена инверсионная вольтамперограмма образца речной воды, содержащего медь, свинец, кадмий и цинк.
45

Рис. 22. Инверсионная вольтамперограмма образца речной воды
Благодаря низкому пределу аналитического обнаружения (10–9 моль/л) инверсионная вольтамперометрия широко используется в анализе природных объектов и особо чистых веществ.
2.6.АМПЕРОМЕТРИЧЕСКОЕ ТИТРОВАНИЕ
Впроцессе развития полярографии был разработан косвенный метод физико-химического анализа, названный амперометрическим титрованием. Он основан на измерении изменения предельного (диффузионного) тока определяемого компонента, титранта или продукта их взаимодействия при титровании.
Амперометрическое титрование может производиться с однимилидвумяиндикаторными электродами.
Для реализации амперометрического титрования с одним поляризованным электродом используют двухэлектродные ячейки, включающие электроды сравнения и индикаторный. Принципиальная схема установки не отличается от схемы установки для постоянно-токовой полярографии (см. рис. 12).
Дополнительным элементом в этой схеме выступает бюретка с раствором титранта.
Для амперометрического титрования используют известные типы химических реакций: окисления-восстановления, оса-
46

ждения, комплексообразования. Для реализации метода необходимо знать электрохимические характеристики веществ, участвующих в химическом взаимодействии. На основании этих данных выбирают потенциал, отвечающий площадке предельного тока участника или продукта реакции, концентрация которого меняется в процессе титрования.
В качестве индикаторных электродов используют вращающийся платиновый или графитовый электроды, а также ртутный капающий. Известно использование в качестве электродного материала углеситалла и стеклоуглерода. Выбор электрода определяется электродной реакцией, используемой при титровании. В этом случае учитывают электродные процессы, связанные с выделением водорода (катодная область) или кислорода (анодная область) (рис. 23).
Рис. 23. Вольтамперные кривые выделения водорода (катодная область) и кислорода (анодная область) на ртутном капающем (1), вращающемся платиновом (2) и графитовом (3) электродах
в 1 М растворе серной кисоты
Как видно, ртутный капающий электрод можно использовать в основном для катодных процессов. На платиновом и графитовом электродах возможны и анодные, и катодные процес-
47
сы. Можно сказать, что эти электроды не заменяют, а дополняют друг друга.
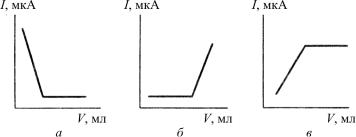
Проведению титрования предшествует вольтамперометрическое исследование для установления условий титрования. Подбирают электрод, на котором электроактивен хотя бы один из участников взаимодействия, и устанавливают потенциал, отвечающий площадке предельного (диффузионного) тока этого участника. Кривые титрования будут иметь различный вид (рис. 24). Так, например, при титровании свинца (II) раствором сульфата калия для установления к.т.т. используют реакцию восстановления определяемого компонента на ртутном капающем электроде. С уменьшением концентрации свинца (II) в растворе по мере добавления титранта ток также будет уменьшаться, а затем достигнет постоянной величины (рис. 24, а).
Рис. 24. Формы кривых амперометрического титрования для случаев, когда активен определяемый компонент (а), титрант (б) и продукт реакции (в)
Если электроактивным является титрант, то ток начнет возрастатьпосле к.т.т. (рис. 24, б). Такую кривуюможно получитьпри титровании цинка (II) раствором K4Fe(CN)6, окисляющемся на платиновом вращающемся электроде. Титрование возможно также, если электрохимически активен продукт реакции (рис. 24, в). Примером являетсятитрованиеарсенат-иона йодидом:
AsO43– + 2 I– + 4 H+ → AsO2– + I2 + 2 H2O.
48
Образующийся продукт реакции I2 электрохимически может восстанавливаться на платиновом электроде:
I2 + 2е– = 2 I–.
Заслуживает внимания применение амперометрического индикатора для случая, когда все участники реакции электрохимически неактивны. Способ заключается в установлении к.т.т. по изменению предельного тока амперометрического индикатора, специально добавляемого к аналиту. Этот индикатор должен реагировать
ститрантомпослезавершенияреакциисопределяемымвеществом. Например, алюминий (III) можно титровать фторид-ионами
сплатиновым вращающимся электродом в присутствии индика-
тора – железа (III). В процессе титрования сначала связываются в комплекс ионы алюминия, фторидные комплексы которого значительно прочнее фторидных комплексов железа (III). После того как алюминий (III) будет полностью связан фторид-ионами, начнется образование фторидных комплексов железа (III), что вызовет уменьшение предельного тока этого индикатора.
Типичныепримерытакоготитрованияприведеныв табл. 10. При простом аппаратурном оформлении метод характеризуется пределом аналитического обнаружения ~10–5 моль/л. Относительное стандартное отклонение результатов составляет 0,01–0,03. Амперометрическое титрование можно использовать в анализе мутных и окрашенных растворов. Его применяют в анализе минерального сырья и продуктов переработки, природных и сточных
вод, фармацевтических препаратов, полимеров и другого. Существует разновидность амперометрического титрова-
ния, при котором используют два одинаковых индикаторных электрода, чаще всего платиновых или графитовых. В иностранной литературе метод известен под названием dead stop and point (метод мертвой конечной точки или метод резкой конечной точки). В соответствии с рекомендациями ИЮПАК за методом установилось название, предложенное И. Кольтгофом, «Амперометрическое титрование с двумя индикаторными электродами».
49

Таблица 1 0
Использование амперометрического титрования с одним индикаторным электродом
Опреде- |
Титрант |
Фон |
|
Элек- |
Потен- |
Электро- |
ляемый |
|
активное |
||||
ион |
|
|
|
трод |
циал, В |
вещество |
|
|
Реакции осаждения |
|
|
|
|
Ва2+ |
K2СrO4 |
0,1 М NAOH |
|
р.к.э. |
–1,0 |
СrO42– |
SO42– |
Pb(NO3)2 |
0,1 МСН3СООН |
|
р.к.э. |
–0,8 |
Рb2+ |
МО42– |
|
0,1 МCH3COONa |
|
|
|
|
Аl3+ |
8-Оксихинолин |
Ацетатный буферный |
|
р.к.э. |
–1,45 |
8-Оксихинолин |
Zn2+ |
|
раствор, рН= 3,5 |
|
|
|
Fe(CN)64– |
K4Fe(CN)6 |
0,1 М K2SO4 |
|
Pt |
+1,0 |
||
|
|
Реакции комплексообразования |
|
|
||
Zn2+ |
ЭДТА |
Ацетатный буферный |
|
р.к.э. |
–1,4 |
Zn2+ |
Bi3+ |
|
раствор, рН= 4,7 |
|
|
|
Bi3+ |
ЭДТА |
ТартратNa, рН= 2 |
|
р.к.э. |
–0,2 |
||
Fe3+ |
ЭДТА |
0,1 МНСl |
|
Pt |
+0,9 |
ЭДТА |
|
|
Реакции окисления-восстановления |
|
|
||
Fe2+ |
K2Cr2O7 |
1 М НСl |
|
Pt |
+1,0 |
Fe2+ |
Fe2+ |
Сl4+ |
1 МH2SO4 |
|
Pt |
±1,2 |
Cl4+ |
Fe3+ |
Аскорбиновая |
1 МH2SO4 |
|
Pt |
+0,95 |
Аскорбиновая |
|
кислота |
|
|
|
|
кислота |
На рис. 25 приведена схема установки для реализации этого метода. Два одинаковых платиновых электрода 1 погружают в анализируемый раствор, перемешиваемый мешалкой 2, и соединяют их через гальванометр (микроамперметр) 3 с регулируемым при помощи реостата 5 источником постоянного тока 6. Разность потенциалов устанавливают с помощью вольтметра 4. При выполнении анализа регистрируют изменение тока в зависимости от объема добавленного титранта. Конечную точку определяют по резкому возрастанию или уменьшению тока, а также по минимуму на V-образной кривой.
Характер кривых титрования зависит от обратимости титруемой системы или системы титранта и от приложенной к электродам разности потенциалов.
50