

Tomchuk_POSІB_VET_BІOHІMІJa
.pdfзастосовуючи в якості індикаторного електроду калій-селективний електрод, а як електрод порівняння – насичений хлорсрібний.
За отриманими даними будують графік в координатах ЕРС – lgСК.
2. Приготування і вимірювання досліджуваного розчина
Розраховують масу наважки препарату, виходячи з міркувань, що вміст калію повинен бути в досліджуваному розчині в межах концентрацій 10-1–10-5 моль/дм3. Точну наважку досліджуваної проби переносять в мірну колбу і розчиняють у розчиннику, який попередньо застосовувався, доводять об’єм колби до мітки і перемішують. Вимірюють ЕРСХ досліджуваного розчину.
Проведення розрахунків:
За виміряним значенням ЕРСХ і побудованим градуювальним графіком розраховують молярну концентрацію іонів калію спочатку у вимірюваному розчині, а потім, враховуючи розведення і масу наважки, розраховують вміст калію в грамах на грам препарату.
Лабораторна робота 2.4. Потенціометричне визначення рН розчинів
У мірну хімічну склянку ємністю 50 см3 поміщають 15–25 см3 досліджуваного розчину, занурюють електроди і вимірюють значення рН або потенціалу системи. При вимірюванні рН розчинів як індикаторний застосовують скляний електрод, а як електрод порівняння – хлорсрібний. Обов’язково контролюється температура розчину.
Порівнюють виміряне значення рН розчину згідно з представленими в табл. 2.6.
Таблиця 2.6. – Значення величини рН досліджуваних розчинів
Назва препарату |
Межі значень рН |
Виміряне значення рН |
Глюкоза, розчин 5, 10, 25, 40 % |
3,0–4,0 |
|
Натрію хлорид (ізотонічний) |
5,0–7,0 |
|
|
|
|
Магнію сульфат 20, 25 % |
6,2–8,0 |
|
|
|
|
Кальцію глюконат 10 % |
6,0–7,5 |
|
|
|
|
Кальцію хлорид 10 % |
5,5–7,0 |
|
|
|
|
101
Лабораторна робота 2.5. Визначення вітаміну А і каротиноїдів
Метод заснований на лужному гідролізі та екстракції вітаміну A і каротиноїдів із подрібненої тканини за допомогою слабко летучих розчинників та наступному спектрофотометричному вимірі поглинання світла розчином до і після руйнування вітаміну A ультрафіолетовими променями при довжині хвилі 328 і 460 нм для каротиноїдів.
Матеріал для дослідження: печінка, пташині яйця. Устаткування, реактиви, лабораторний посуд і матеріали:
спектрофотометр; ультрафіолетова лампа ПРК-4; центрифуга; водяна баня; вентилятор настільній; пробірки зі скла пірекс, що пропускають ультрафіолетові промені, 55х8 мм; центрифужні пробірки; піпетки градуйовані на 1 см3, 5, 10 см3; скляні палички; 96 % спирт етиловий, ксилол або про-ксилол, октан, калію гідрок-сид, 1 н. розчин калію гідроксиду на 96-% етиловому спирті.
Приготування розчинів:
1) До 1 об’єму 11 н. розчину калію гідроксиду (11 н. розчин калію гідроксиду: 617,16 мг калію гідроксиду доводять дистильованою водою в колбі на 1 дм3) додати 10 об’ємів етилового спирту. Розчин готується в день проведення аналізів.
2) Ксилоло-октанова суміш (1:1). Готують у день проведення аналізів.
Хід роботи:
Для дослідження використовують свіжу печінку абу ту, що зберігалася в замороженому стані. Пробу печінки ретельно розтирають у порцеляновій ступці, потім зважують 0,5 г отриманої гомо - генної маси (у такий же спосіб готують для аналізу пробу жовтка, тільки жовток у кількості 1 г відважують безпосередньо в центрифужну пробірку).
Наважку печінки кількісно переносять у центрифужну пробірку і доливають 2 см3 1 н. спиртового розчину калію гідроксиду. Перемішують скляною паличкою до утворення однорідної суміші і ставлять для гідролізу на водяну баню при температурі 60 °C на 30 хв. Після цього пробірки охолоджують у холодній воді протягом 5–10 хв і додають у кожну 8 см3 ксилоло-октанової суміші. Пробірки закривають пробками і сильно струшують протягом 2 хв, після чого центрифугують 5 хв при 1500 об/хв. Верхній шар центрифу-
102
гату переносять піпеткою в кварцову кювету спектрофотометра і колориметрують: каротиноїди визначають при довжині хвилі
460 |
нм. Вітамін A шляхом дворазового виміру, до і після опромі- |
|||
нення |
проб ультрафіолетовими променями, при довжині хвилі |
|||
328 |
нм. |
Для цього досліджувані проби |
переносять з |
кювет у |
пробірки зі скла пірекс і опромінюють 45 |
–60 хв лампою |
ПРК-4 на |
||
відстані 15–19 см. Для того щоб пробірки не нагрівалися, під час опромінення їх охолоджують за допомогою настільного вентилятора. Концентрацію вітаміну A визначають за різницею екстинцій при спектрофотометруванні до і після опромінення.
Проведення розрахунків:
Х= 4,8 · Е · 2 · n,
Х– вміст каротиноїдів, мкг/г;
4,8 – коефіцієнт для каротину; Е – оптична щільність проби при 460 нм;
2 – коефіцієнт перерахунку на 1 г печінки;
n – розведення (кількість мл ксилоло-октановой суміші).
Визначення змісту вітаміну A виконують за формулою:
Х = 6,37 · (Е1 – Е2 ) · 2 ·n, де
Х – кількість вітаміну A, мкг/г; 6,37 – коефіцієнт для вітаміну A;
Е1 – оптична щільність розчину вітаміну A до опромінення при
328 нм;
Е2 – оптична щільність розчину вітаміну A після опромінення при 328нм;
2 – коефіцієнт перерахунку на 1 г печінки;
n – розведення (кількість мл ксилоло-октановой суміші).
Примітка: При високому вмісті вітаміну A в печінці потрібно проводити додаткове розведення, що враховують у формулі розрахунку.
103
Лабораторна робота 2.6. Визначення вмісту хімічних елементів методом атомно-емісійної спектрометрії
Суть роботи полягає у визначенні вмісту хімічних елементів
(Al, B, Ba, Ca, Cd, Co, Cr, Cu, Fe, K, Mg, Mn, Na, Ni, Pb, Sr, Zn, Se,
As) методом атомно-емісійної спектрометрії з індуктивно-зв’язаною плазмою.
Матеріал для дослідження: м’ясо.
Устаткування, реактиви, лабораторний посуд і матеріали:
атомно-емісійний спектрометр з індуктивно-зв’язаною плазмою; пробірки пластикові місткістю 10 см3; колби мірні скляні місткістю 100 см3; дозатори автоматичні одноканальні: дозатор з регульованим об’ємом від 1 до 10 см3, дозатор з регульованим об’ємом від 1 до 5 см3, дозатор з регульованим об’ємом від 0,1 до 1 см3; склянки хімічні 50 см3; кислота нітратна 69 %; гідрогену пероксид 30 %; вода деіонізована; багатоелементні стандартні розчини фірми
MERK.
Хід роботи:
Наважку масою 2 г поміщають в тефлоновий стакан мікрохвильової печі. Додають 7 см3 концентрованої азотної кислоти та 2 см3 гідрогену пероксиду. Залишають на дві години. Герметично закривають і ставлять у мікрохвильову піч для кислотного розкладання, згідно інструкції. Відкривають тефлонові стакани та переносять пробу у пластикову пробірку місткістю 10 см3. Додають 0,4 см3 розчину скандію (50мг/дм3) і доводять до мітки деіонізованою водою. Проби розводять у 5 разів.
Приготування стандартних розчинів:
1) 1 см3 багатоелементного стандартного розчину (М32/10сh) фірми Merk з концентрацією 1000 мг/дм3 доводять до 10 см3 (кінцева концентрація 100 мг/дм3);
2) у три скляні мірні колби місткістю 100 см3 додають по 4 см3 розчину скандію (50мг/дм3); у колбу № 1 до мітки доводять 5 % розчином нітратної кислоти – це нульовий стандарт; у колбу № 2 вносять 1 см3 багатоелементного розчину (концентрація 100 мг/дм3) і доводять до мітки 5 % розчином нітратної кислоти – це стандартний розчин 1 мг/дм3; у колбу № 3 вносять 5 см3 багатоелементного розчину (концентрація 100 мг/дм3) і доводять до мітки 5 % розчином нітратної кислоти – це стандартний розчин 5 мг/дм3;
104
Перед початком вимірювання треба проаналізувати багатоелементний стандартний розчин з найбільшим вмістом елементів як невідому пробу. Переконуються, що відхилення визначеного вмісту від істинних значень не перевищують ±5 % (чи нижчих заданих меж). Вимірювання починають з промивання системи неробочим розчином з реагентом та промивають її після кожної проби. Щоб перевірити міжелементні та фонові чинники корекції, аналізують контрольну пробу для засобу вимірювання перед початком, наприкінці та періодично під час аналізування проб. Результати аналізу мають бути в межах двох стандартних відхилень середнього значення. У іншому випадку припиняють аналіз, усувають проблему та перекалібровують прилад.
Хід роботи:
1.Відкривають потік газу (аргон – об’ємна частка аргону, min
%99,999) та продувають прилад 30–60 хв згідно з інструкцією на
прилад.
2.Запускають на комп’ютері програму TIVA. На приладі встановлюють правильні робочі параметри, як указано в інструкції.
3.Підпалювання плазми і стабілізація її 10–15 хв.
4.Вводять багатоелементний розчин і розташовують МАСКИ елементів (місця).
5. Виводять калібрувальні стандартні розчини 0 мг/дм3; 1,
5мг/дм3.
6.Вводять калібрувальний стандартний розчин (1 мг/дм3) як пробу для перевірки відтворюваності.
7.Проводять аналіз зразків.
Систему промивають деіонізованною водою після кожного контрольного зразка та промивають її після кожної проби. Через кожні 10 проб аналізують стандарт з відомою концентрацією елементів. Результати представляють в міліграмах на кілограм (мг/кг), або грам на кілограм (г/кг).
Лабораторна робота 2.7. Визначення показника заломлен-
ня
Показник заломлення характеризує чистоту, ненасиченість, ступінь окислення жирів. Показник заломлення зростає за наявності оксигруп, збільшенні молекулярної маси і кількості неграничних
105
жирних кислот, що входять до складу жиру. Зміна температури приводить до зміни щільності речовини.
З підвищенням температури на 1 °С щільність знижується в середньому на 0,00037. Отже, показник заломлення зменшується. Для жирів показник заломлення визначають при температурі 20 °С або шляхом розрахунку приводять до 20 °С.
По величині показника заломлення можна судити про природу жиру, його чистоту і ступені окислення. У окисленому жирі показник заломлення вище в порівнянні з показником заломлення свіжого жиру внаслідок збільшення молекулярної маси (унаслідок утворення оксигруп тощо).
Матеріал для дослідження: жири тваринні і олії рослинні.
Устаткування, реактиви, лабораторний посуд і матеріали:
рефрактометр для вимірювання показника заломлення від 1,3000 до 1,7000; термометр; етер етиловий лауринової кислоти для рефрактометрії, з відомим показником заломлення; гексан або петролейний етер.
Хід роботи:
Показник заломлення визначають на зневодненому та профільтрованому жирі. Тверду пробу переносять в склянку, переносять у водяну баню, що налаштована на температуру, за якої буде виконуватися вимірювання, та витримують доти, поки проба повністю не стабілізується.
Перевіряють калібровальну криву рефрактометра за допомогою вимірювання показника скляної пластинки у відповідності до інструкції виробника чи вимірюючи показник заломлення етилового етеру лауринової кислоти.
Пробу дослідного жиру ретельно перемішують та профільтровують. Перед визначенням показника заломлення поверхні призм рефрактометра протирають м’якою тканиною з бавовни, яка оброблена гексаном чи петролейним етером. Рефрактометр готують до використання згідно інструкції, яка додана до приладу. Перевірку рефрактометра та корекцію нуля проводять згідно з інструкцією.
Відповідно до інструкції визначають показник заломлення дослідного зразка при 20 ºС, 40, 50, 60 і 80 °С, залежності від того, за якої температури проба рідка. Вимірювання проводять три рази. Поверхню призми одразу після вимірювання витирають тканиною,
106

а потім ватою, яка оброблена гексаном чи петролейним етером і дають висохнути.
Проведення розрахунків:
Якщо різниця між фактичною температурою ( ), за якої проводилося вимірювання, і раніше заданої
), за якої проводилося вимірювання, і раніше заданої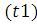 менше 3 °С, показник заломлення розраховується за формулою:
менше 3 °С, показник заломлення розраховується за формулою:
 – показник заломлення при температурі досліду; t – температура досліду, °С;
– показник заломлення при температурі досліду; t – температура досліду, °С;
t1 – завдана температура (20, 40, 50, 60, 80), °С;
F – коефіцієнт, що має значення:
0,00035 – за t = 20°С,
0,00036 – за t = 40 ºС, 50, 60 °С, 0,00037 – за t = 80°С.
Якщо різниця між фактичною температурою (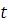 ), за якої проводилося вимірювання, і раніше заданої
), за якої проводилося вимірювання, і раніше заданої  дорівнює чи перевищує
дорівнює чи перевищує
3 °С, тоді виконують нове визначення.
За кінцевий результат беруть середнє арифметичне трьох паралельних визначень. Визначення проводять до четвертого знаку після коми. Різниця між двома паралельними визначеннями не повинна перевищувати 0,0002.
Лабораторна робота 2.8. Визначення жирнокислотного складу тваринних жирів
Однією з основних ідентифікаційних характеристик жирів є їх жирнокислотний склад. Саме по ньому можна виявити фальсифікацію жирів і маргаринової продукції. Жирнокислотний склад визначається методом газової хроматографії. За допомогою цього методу визначають якісний і кількісний жирнокислотний склад, кількість транс-ізомерів.
Метод базується на перетворенні триацилгліцеролів жирних кислот в метилові етери жирних кислот та газохроматографічному аналізі метилових етерів. Метод використовується в діапазоні 0,1–
100 %.
Матеріал для дослідження: жири тваринні і олії рослинні.
107
Устаткування, реактиви, лабораторний посуд і матеріали:
газовий хроматограф з полум’яно-іонізаційним детектором і програмним забезпеченням температури; високополярна капілярна колонка, наприклад SPTM – 2560, 100m x 0.25mm ID, 0.20μm film (Supelco); мікрошприц МШ-10 на 10 мкдм3; аналітичні ваги, з точністю до 0,001 г; генератор водню; колба конічна на 100 см3; зворотний холодильник; воронка лабораторна; піпетки мірні на 10 см3; віали з темного скла на 4 см3; гексан; натрій хлористий, насичений розчин; натрію сульфат, зневоднений; натрію гідроксид, метиловий розчин 0,5 н.; бору трифторид, метиловий розчин; ізооктан; стандартна суміш метилових етерів жирних кислот, наприклад Supelco TM 37 Compone FAME Mix, 100mg Neat; гелій газоподібний стис-
нений 99,999%.
УВАГА! Метод, який описано, передбачає використовування потенційно небезпечних реактивів. Треба дотримуватися звичай - них заходів техніки безпеки щодо захисту очей і захисту від хімічних опіків. Бору трифторид отруйний.
Хід роботи:
Дослідну пробу (100–250 мг) ретельно перемішують та вміщують у колбу 50 см3. Додають 4 см3 метанольного розчину натрію гідроксиду. Зворотний холодильник приєднують до колби і кип’я- тять зі зворотним холодильником до зникнення крапельок жиру, обережно помішуючи вміст колби з інтервалом від 30 с до 1 хв, щоб запобігти формуванню кільця з натрію гідроксиду на стінках колби. Ця процедура, звичайно, потребує від 10 хв, але іноді до 1 год. Додають 5 см3 метанольного розчину бору трифториду через верхню частину холодильника. Продовжують кип’ятіння протягом від 30 до 60 хв. Додають у киплячу суміш через верхню частину холо - дильника від 1 до 3 см3 гексану (ізооктану). Знімають колбу з джерела тепла та від’єднують зворотний холодильник.
НЕГАЙНО, не даючи колбі охолонути, додають 20 см3 розчину натрію хлориду. Колбу закривають корковою пробкою і інтенсивно струшують протягом 15 с. Додають ще насиченого розчину натрію хлориду, щоб довести рівень суміші до горловини колби. Дають двом фазам можливість розділитися. Від 1 до 2 см3 верхньоого гексанового шару переносять у флакон місткістю 4 см3 і додають невелику кількість зневодненого натрію сульфату для повного видалення слідів води.
108

Проводять аналіз метилових етерів жирних кислот методом газової хроматографії.
Хід роботи:
Включають газовий хроматограф в електромережу. Встановлюють параметри аналізу в приладі згідно інструкції на колонку і параметрам описаних в методиці. Дають приладу вийти на режим і прогрітися 30 хв.
Проводять холостий дослід з н-гептаном або гексаном. В холостому досліді не повинно бути знайдено ніяких піків, крім розчинника. Це випробування повторюють після кожних десяти проб.
Перед проведенням аналізу метилових етерів жирних кислот дослідного зразку, проводять хроматографічний аналіз стандартної суміші метилових етерів жирних кислот (Supelco TM 37 Compone FAME Mix, 100mg Neat). Ця процедура необхідна для того, щоб перевірити чутливість та ефективність розділення хроматографічної колонки, а також для проведення ідентифікування отриманих хроматографічних піків компонентів проби.
Потім проводять аналіз метилових етерів жирних кислот досліджуваного зразка. Для цього використовуючи мікрошприц, відбирають 1 мкдм3 розчину метилових етерів жирних кислот і вводять у колонку газового хроматографа. Проходить хроматографічний аналіз. Після проведення аналізу прилад записує хроматограму поділу компонентів. З графіка визначають піки метилових етерів. Ідентифікацію проводять за допомогою стандартних зразків метилових етерів жирних кислот.
Проведення розрахунків
Використовують метод внутрішньої нормалізації, тобто вважають, що сума площин усіх піків компонентів проби (ΣSi), які представлені на хроматограмі, становить 100 %. Площа піків компонентів (S) в квадратних міліметрах розраховується за формулою:
hi – висота піку, мм;
ai – ширина, яка виміряна на половині висоти, мм.
Масову частку жирної кислоти у відсотках розраховують за формулою:
109

S – площа піку метилового етеру жирної кислоти, мм2, ΣSi – сума площин усіх піків на хроматограмі, мм2.
Розрахунки проводять до другого десяткового знаку з наступним округленням результату до першого десяткового знаку. За результат аналізу приймають середнє арифметичне результатів двох послідовних визначень.
Лабораторна робота 2.9. Визначення вмісту афлатоксинів
Афлотоксини – токсичні та канцерогенні речовини, що виробляються різними видами грибів роду Aspergillus. Серед всіх токсинів, які виробляються біологічним шляхом, афлотоксини – найбільш токсичні. Існує три основні групи афлатоксинів:
Афлатоксини групи В: входять афлатоксини В1 (найпоширеніший, найбільш токсичний та канцерогенний) та В2.
Афлатоксини G: в цю групу входять афлатоксини G1 та G2; Афлатоксини М: в цю групу входять афлатоксини М1 та М2. Це
метаболіти афлатоксину B1, які виявляються у молоці корів.
Матеріал для дослідження: зерно.
Устаткування, реактиви, лабораторний посуд і матеріали:
рідинний хроматограф високого тиску з флуоресцентним детектором; ваги лабораторні; орбітальний шейкер; дистильована або деіонізована вода; маніфолд з вакуумним насосом; натрію хлорид; целіт (для жирових продуктів); хлороформ (для жирових продуктів); фосфатний буфер розчин (PBS) таблетки; фільтрувальний папір (синя стрічка) та все необхідне для фільтрування; піпетки, об’єм дозування 1–10 см3; лабораторні стакани, 250 см3; конічні колби, 250 см3; конічні колби, 500 см3 (для жирових продуктів); мірні колби, 250 см3; пробірки, 5–10 см3; ацетонітрил; метанол; стандартний розчин афлатоксинів В1, В2, G1, G2.
Хід роботи
Зважити 50 г мілко подрібненої проби та 4 г натрію хлориду, помістити в літрову ємність блендеру (стійкого до розчинників). Додати 250 см3 метанол:дистильована вода (60:40) накрити і перемішувати 1 хв при високій швидкості (на шейкері протягом 15 хв). Розвести екстракт у 250 см3 дистильованої води. Обережно і добре перемішати (вручну). Негайно після перемішування, відфільтрувати
110