

Глава 2 нейрофармакология опиатов и опиоидов фармакодинамика и фармакокинетика наркотических анальгетиков
Классическим представителем наркотических анальгетиков — опиатов — является морфин, выделенный из опийного мака еще н 1803 г. Морфин — основной алкалоид опийного мака, где его содержание колеблется от 3 до 23 %. На основании структуры морфина, а также путем направленного синтеза были созданы новые высокоактивные болеутоляющие средства и их фармакологический антагонист налоксон. В последние годы уточнены терминологические критерии, согласно которым болеутоляющие соединения, содержащиеся в соке опийного мака, принято называть опиатами, а вещества другого химического строения, близкие по фармакологическим эффектам к опиатам, — опиоидами или опиатоподобными веществами.
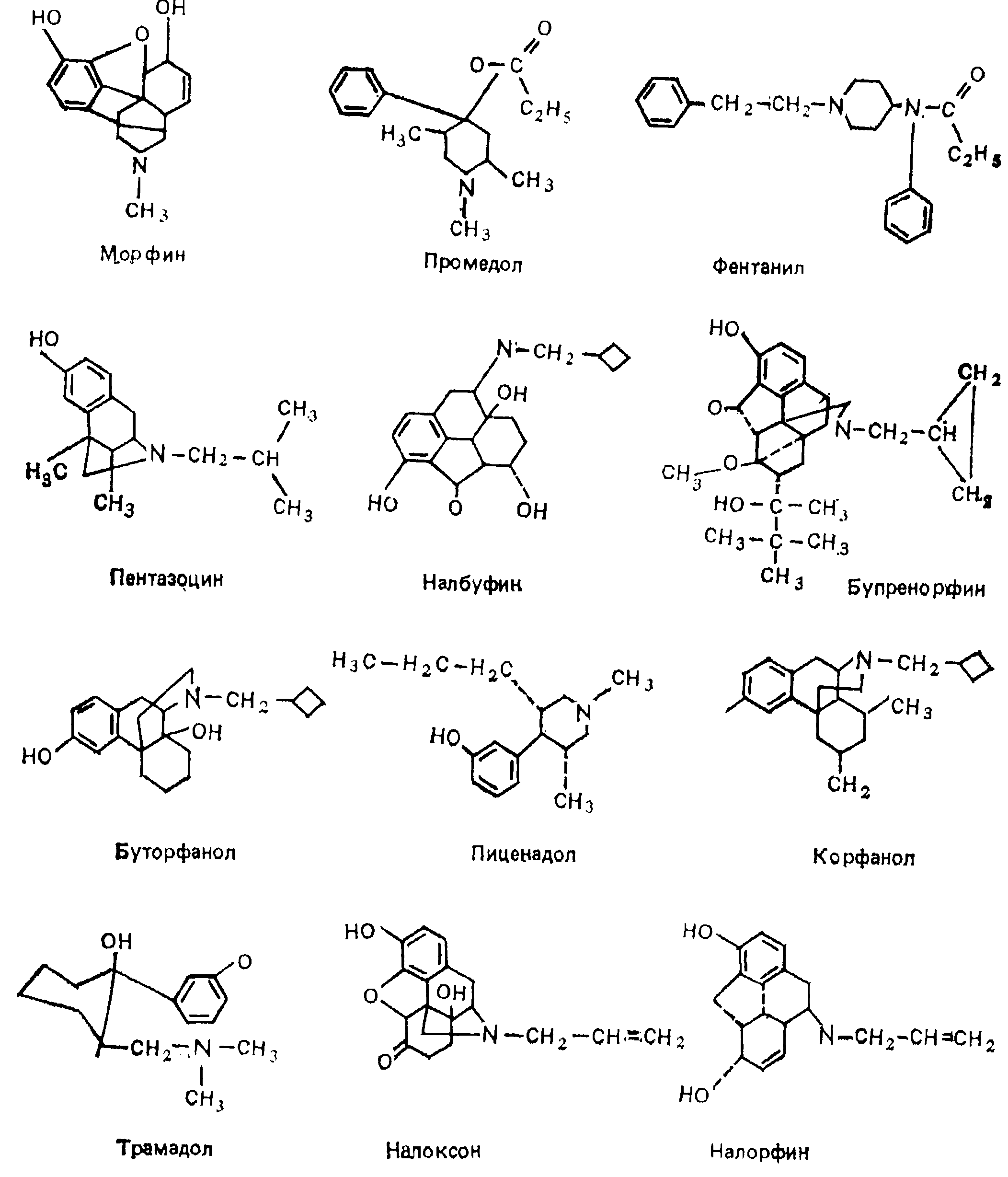
По избирательности и характеру их влияния на опиатные рецепторы наркотические анальгетики разделяются на несколько групп: морфиноподобные агонисты — морфин и его производные (гидроморфин, оксиморфин и др.), промедол (меперидин, демерол), фентанил и его производные (суфентанил, альфентанил, карфентанил и др.), метадон (долофин), леворфанол (леводроморан), эторфин, трамадол (трамал), а также сравнительно редко использующиеся кодеин и героин; смешанные агонисты — антагонисты — пентазоцин (лексир, фортрал, талвин), буторфанол (стадол), налбуфин (нубаин), налорфин, корфанол, пиценадол; частичные (парциальные) агонисты — бупренорфин (темгесик). Детальная характеристика традиционных и новых наркотических анальгетиков дана в ряде обобщающих работ последних лет [Осипова Н.А., 1988; Davis P., Cook D., 1986; Mehlich D., 1986; Freye E, 1987; Hare В., 1987, и др.].
Морфин. Большой опыт клинического применения морфина показал, что препарат обладает несомненными достоинствами: обеспечивает глубокое обезболивание, не сопровождающееся амнезией, не вызывает сенсибилизации миокарда к катехоламинам, не нарушает регуляции кровотока в головном мозге, сердце, почках, не оказывает токсического действия на печень, почки и др. Вместе с тем морфин нельзя признать идеальным анальгетиком прежде всего в связи с его высоким наркогенным потенциалом, способностью угнетать дыхание, вызывать обстипацию и некоторыми другими свойствами. Недостатком морфина является сравнительно низкая биодоступпость при приеме внутрь, что находит отражение в разнице эквианальгетических доз при энтеральном и внутримышечном способе введения. Последний путь, наряду с подкожным введением, обеспечивает оптимальную длительность действия морфина, тогда как после его внутривенного введения период полувыведения (T1/2) составляет всего около 100 мин. Морфин частично связывается с белками плазмы. Пороговое анальгетическое действие развивается при концентрации свободного морфина в плазме крови 30 нг/мл. Лишь незначительная часть от введенного морфина (менее 0,01 %) обнаруживается в ткани головного мозга, что, вероятно, связано с относительно низкой липоидотропностью препарата. Выводится из организма морфин главным образом через почки, преимущественно в виде глюкуронида. Данные о кинетике морфина у больных с почечной недостаточностью противоречивы: отмечают как ее практическую неизменность, так и почти 10-кратное замедление выведения морфина [Sawe J. et al., 1985; Woolner D. et al., 1986]. В то же время отчетливо выявлены зависимость фармакокинетики морфина от возраста и достоверное повышение концентрации свободного морфина после 60 лет, чем объясняется большая чувствительность к препарату пожилых лиц. Имеют значение и другие факторы, пока не изученные в условиях клиники. Так, в экспериментах установлено, что активность морфина может изменяться в 7 раз в зависимости от времени суток и от фазы менструального цикла [Papri В. et а1., 1983].
Высокий наркогенный потенциал ограничивает длительное (за исключением инкурабельных больных) применение морфина. К сожалению, уже при его 1—2-кратном введении проявляется большое число побочных реакций, среди которых наиболее выражены угнетение дыхания, тошнота и рвота, спазмы гладкомышечных органов. Примечательно, что негативные эффекты морфина прямо коррелируют с его концентрацией в крови.
Промедол — отечественный синтетический аналог меперидина, примерно в 5—6 раз менее активен, чем морфин, при различных способах введения. Обладает сходной с морфином фармакокинетикой и, соответственно, длительностью болеутоляющего действия, в эквианальгетических дозах отчетливо угнетает дыхание. Обычно используется при болевых синдромах средней выраженности в небольших дозах (около 40 мг на 70 кг массы тела парентерально), что минимизирует депрессию дыхания и практически нивелирует изменение тонуса гладкомышечных органов. Более того, промедол дает некоторый родостимулирующий эффект, благоприятно влияет на кровообращение в беременной матке, что позволяет рассматривать его как средство выбора в акушерской клинике [Куликов В.И., Меркулова Е.В., 1984].
Фентанил. Открытие в период с 1957 по 1962 г. новых наркотических анальгетиков и среди них фентанила привело к существенному изменению роли опиоидов в анестезиологическом пособии. Именно фентанил явился основой таких новых способов обезболивания, как нейролептаналгезия, синаптоаналгезия, атараналгезия. В конце 60-х годов фентанил вместе с морфином стал применяться в больших дозах в качестве основного или единственного компонента наркоза. Фентанил отличается очень высокой болеутоляющей активностью, однако резко угнетает дыхание, особенно у пожилых лиц, вызывает ригидность дыхательной мускулатуры и мышц брюшной стенки.
Вводится фентанил преимущественно внутривенно или внутримышечно, при этом скорость развития обезболивающего эффекта составляет 1—3 мин и 10—15 мин соответственно, а продолжительность аналгезии не превышает 30 мин. Быстрая и выраженная аналгезия обусловлена высокой липоидотропностью фентанила и его способностью хорошо проникать через гемато-энцефалический барьер. Основными органами метаболизма являются печень и в значительно меньшей степени — почки, в которых осуществляются окислительное дезалкилирование и гидроксилирование фентанила до фенилуксусной кислоты, норфентанила и некоторых других продуктов, которые вместе с небольшой фракцией неизмененного фентанила выделяются с мочой. В настоящее время нашли применение ряд аналогов фентанила: альфентанил, суфентанил, лофентанил, — последний из которых обладает наибольшей продолжительностью действия [Davis P., Cook D., 1986]. Для фентанила и ею производных характерно брадикардическое действие, обусловленное, по-видимому, активацией центральных парасимпатических механизмов, поскольку брадикардия предупреждается атропином. Другие побочные эффекты, выраженные у эталонного анальгетика морфина, при применении фентанила и его аналогов наблюдаются редко. Более того, по данным экспериментов, у суфентанила необычайно высок терапевтический индекс (отношение ЛД50/ЕД50). который равен 26716 [Freye E., 1987]. Терапевтический индекс морфина 71.
Пентазоцин — синтетический анальгетик, один из наиболее хорошо изученных представителей нового класса опиоидов, обладающих смешанным агонист-антагонистическим взаимодействием с опиатными рецепторами. Современными методами исследования доказана несостоятельность предположения о том, что особенности фармакологических эффектов пснтазоцина обусловлены его селективностью к х-опиатным рецепторам. Сравнительно большая избирательность влияния на х-рецепторы характерна для тифлуадома, бремазоцина, который хорошо проникает в мозг при парентеральных способах введения. По анальгетической активности пентазоцин в 3—6 раз слабее морфина, однако в анальгетических дозах вызывает такую же депрессию дыхания, активирует центральные симпатические механизмы, вследствие чего развиваются гипертензия и тахикардия, может ухудшать коронарный кровоток [Goldstein G., 1985]. Скорость и длительность болеутоляющего эффекта пентазоцина сходны с таковыми при применении морфина. В связи с низкой биодоступностью (20 %) действие пентазоцина при энтеральном применении выражено слабее, чем при внутривенном или внутримышечном способах введения. Кинетика пентазоцина обычно би- или триэкспоненциальная со средним T1/3 3—5ч, плазменным клиренсом 1200—2800 мл/мин и объемом распределения 200—400 л. Выводится пентазоцин из организма почками, преимущественно в виде метаболитов. Достоинствами его являются слабое проникновение через плаценту и благоприятное влияние на сократительную активность миометрня, на чем основано его применение в акушерской практике. Помимо угнетения дыхания, побочные реакции пентазоцина включают психотомиметические, желудочно-кишечные и другие побочные эффекты.
Бупренорфин — производное орипавина, обладает высоким аффинитетом к μ- и х-опиатным рецепторам. Однако его фармакологический профиль первично определяется агонизмом к μ-рецепторам и необычно замедленной кинетикой их освобождения. Эти особенности обусловлены наличием в структуре буиренорфина N-бутильной группировки в положении С7, придающей ему сходство с энкефалинами и определяющей его сильное и длительное болеутоляющее действие [Freedman М., 1986; Salvi E. ei al., 1986]. Бупренорфин считается первым наркотическим анальгетиком, с помощью которого в определенной степени достигнуто «разделение» болеутоляющих и токсикоманических свойств. Он обладает очень высокой, близкой к фентанилу, анальгетической активностью и, в отличие от последнего, высокой биодоступностью, которая колеблется в зависимости от способа введения от 40 до 100 %. При парентеральном введении разовая анальгетическая доза, обеспечивающая достаточный эффект при умеренных и сильно выраженных болевых синдромах, составляет 0,3 — 0,6 мг на 70 кг массы тела, T1/3 составляет от 3 до 5 ч, максимальное анальгетическое действие длится не менее 6 ч.
Выраженность угнетающего действия бупренорфина на дыхание совпадает с таковой для морфина в диапазоне оптимальных анальгетических доз, однако этот нежелательный эффект бупренорфина практически не зависит от дозы. Описано применение бупренорфина без тяжелых последствий в дозе 8 мг/сут в течение нескольких дней подряд. Бупренорфин считается удобным препаратом для терапии послеоперационных болей, причем с этой целью рекомендуется его сублингвальное применение в таблетках (0,2 мг). В этом случае биодоступпость бупренорфина составляет в среднем 55 %, T1/2 — 76 мин при значительной продолжительности действия. Из побочных эффектов отмечают тошноту, рвоту, сонливость, выраженность которых прямо зависит от дозы препарата. Терапевтический индекс бупренорфина (около 8000) значительно выше, чем у пентазоцина (4).
Другие представители этой группы агонист-антагонистов опиатных рецепторов принципиально отличаются от бупренорфина. Они вызывают аналгезию, воздействуя на х-рецепторы, а за счет μ-антагонистической активности препятствуют угнетению дыхания [Freye E., 1987]. К этой группе относятся производные морфина, синтезированные в 80-х годах, — налбуфин, буторфанол, корфанол, пиценадол [подробнее см.: Howes J. et al., 1985; Pachter I., Evens R„ 1985; Zimmerman D. et al., 1985; Schmidt W. et al., 1985]. Все они характеризуются минимальным угнетением дыхания, незначительным влиянием на желудочно-кишечный тракт и сердечно-сосудистую систему. Однако небольшой диапазон между анальгетичсскими дозами и дозами, вызывающими психотомиметические расстройства, ограничивает их широкое применение в клинике.
Налбуфин равен морфину по анальгетической активности при внутримышечном введении, при энтеральном приеме эффективность налбуфина в 4—5 раз ниже. Пик концентрации в плазме крови возникает через 30 — 60 мин, длительность действия 3—6 ч, T1/2 составляет 2—3 и 7—8 ч при парентеральном и энтеральном введении соответственно. Метаболизируется налбуфин в печени и выделяется с желчью через кишечник. Очень незначительная часть неизмененного налбуфина экскретируется с мочой. Наиболее типичный побочный эффект налбуфина — седативное действие, которое возникает у 36 % пациентов. Другие побочные эффекты бывают редко, например, тошнота и рвота — всего в б % случаев. Выраженность угнетения дыхания под влиянием налбуфина в дозе 10 мг (внутривенно) сходна с эффектом морфина в такой же дозе. Однако при увеличении дозы налбуфина депрессия дыхания не усиливается. Налбуфин обладает сравнительно низким психотомиметическим потенциалом, слабым влиянием на моторику желудочно-кишечного тракта, минимальной толерантностью и способностью вызывать физическую зависимость.
Буторфанол по своим свойствам очень близок к налбуфину и отличается от последнего более высокой анальгетической и психотомиметической активностью.
Корфанол — препарат, эффективный при энтеральном применении и обладающий резистентностью к антагонистам опиатов. На основании преимущественно экспериментальных данных предполагают, что корфанол и еще один новый аналыетик — пиценадол — обладают минимальным наркогенным потенциалом.
Трамадол — новый синтетический анальгетик со сравнительно высокой (60—70 %) биодоступностью при разных способах введения, быстрым и длительным болеутоляющим эффектом. Однако он уступает морфину по анальгетической активности в 5—10 раз. После внутривенного введения трамадола болеутоляющее действие развивается через 5—10 мин, T1/3 составляет около 6 ч. При энтеральном введении аналгезия возникает через 30—40 мин и не снижается в течение 9 ч. В обоих случаях используют трамадол в дозах 100 — 200 мг на 70 кг массы тела, что обеспечивает создание в крови анальгетической концентрации — 100 нг/мл и более [Lintz W. et al., 1986]. На фоне трамадола отмечают стабильность параметров кровообращения. К сожалению, препарат не лишен характерных для опиоидов нежелательных эффектов: часто возникают тошнота и рвота, характерным также считают угнетение дыхания в раннем послеоперационном периоде.
У трамадола один из наиболее низких терапевтических индексов, равный 3.
Выбор препарата для использования в конкретной клинической ситуации должен определяться в первую очередь фармако-кинетическими характеристиками, с обязательным учетом особенностей организма (возраст, состояние систем метаболизма).
В табл. 3 приведены сравнительная оценка болеутоляющей активности и длительности действия традиционных и новых наркотических анальгетиков и некоторые показатели их фармакокинетики при энтеральном и парентеральном способах введения
Особое значение имеет знание особенностей фармакодинамики и фармакокинетики опиатов и опиоидов у детей. Эти особенности относятся главным образом к новорожденным, у которых T1/2 большинства анальгетиков пролонгирован. У детей кажущийся объем распределения препаратов увеличен в связи с уменьшением их фиксации белками плазмы, уменьшенной массой липидов и повышенным количеством воды в тканях. Элиминация наркотических анальгетиков снижена в связи с особенностями метаболизма в печени и функции почек, особенностями проникновения отдельных препаратов в ЦНС, которые во многом определяют их активность и токсичность. Показано, что морфин и пентазоцин более токсичны у новорожденных, чем у взрослых, тогда как токсичность промедола и фентанила не зависит от возраста [Bloch R., Belhadj Z., 1983].
Завершая краткое изложение свойств наркотических анальгетиков, следует подчеркнуть парадоксальность ситуации, которая заключается в том, что, даже при наличии определенного набора лекарственных средств, способных эффективно корригировать болевые синдромы практически любой выраженности, неудачи в обезболивании могут достигать 70 % [Slattery P., Boas R., 1985]. Одной из причин этого является неправильное применение анальгетиков — неэффективные дозы, нарушение режима и метода введения и т.п. Очень часто не удается до стигнуть необходимой концентрации апальгетиков в крови и тем более поддерживать ее длительное время. Для совершенствования опиатной аналгезии рекомендуют вводить препараты длительного действия, например бупренорфин, для уменьшения депрессии дыхания — соединения смешанного агонист-антагонистического действия. Создание новых анальгетиков приводит к тому, что у них уменьшается какой-либо побочный эффект, однако проявляется способность вызывать другие нежелательные реакции. Эта закономерность четко прослежена в обзоре Т. Bewley, A. Ghodse (1984), поэтому на современном уровне понимания механизмов действия наркотических анальгетиков становится очевидным, что дальнейший поиск новых аналогов морфина и опиоидов может кардинально улучшить опиатную аналгезию только в случае направленного создания соединений с заданным спектром рецепторного действия.
Т а б л и ц а 3
Сравнительная характеристика наркотических анальгетиков
(по С. Inturrisi, 1982, в собственной модификации)
Препарат (международ-ное название) |
Путь введения |
Эквианальгетичсские дозы (мг) |
Аналгезия, ч |
Т ½ ч |
|
Максимум |
Длительность |
||||
Морфин |
Внутримышечно |
10 |
0,5—1 |
4—6 |
2—3,5 |
|
per os |
60
|
1,5—2 |
4—7 |
|
Меперилин |
Внутримышечно |
75 |
0,5—1 |
4—5 |
3—4 |
|
per os |
300 |
1—2 |
4—6 |
|
Леворфанол |
Внутримышечно |
2 |
0,5—1 |
4—6 |
12—16 |
|
per os |
4 |
1,5—2 |
4—7 |
|
Пентазоцин |
Внутримышечно |
60 |
0,5—1 |
4—6 |
2—3 |
|
per os |
180
|
1,5—2 |
4—7 |
|
Налбуфин |
Внутримышечно |
10 |
0,5—1 |
4—6 |
5 |
Буторфанол |
Внутримышечно |
2 |
0,5—1 0,5—1 |
4—6 |
2,5—3,5 3—5 |
Бупренорфин |
Внутримышечно |
0,4 |
0,5—1 |
6—8 |
|
|
Сублингвально |
0,8 |
2—3 |
6—12 |
|