

3.2.5. Електрофорез білків в агаровому гелі
Подібно до крохмального й акриламідного, агаровий гель є дуже м’яким носієм. На відміну від електрофорезу на папері, в агаровому гелі не відбувається інактивації білків, що дає змогу визначати активність окремих білкових фракцій безпосередньо в гелі після завершення електрофорезу. Приготування агарового гелю є набагато простішим, ніж приготування крохмального чи поліакриламідного гелів. Тривалість електрофорезу в агаровому гелі – 1–4 год.
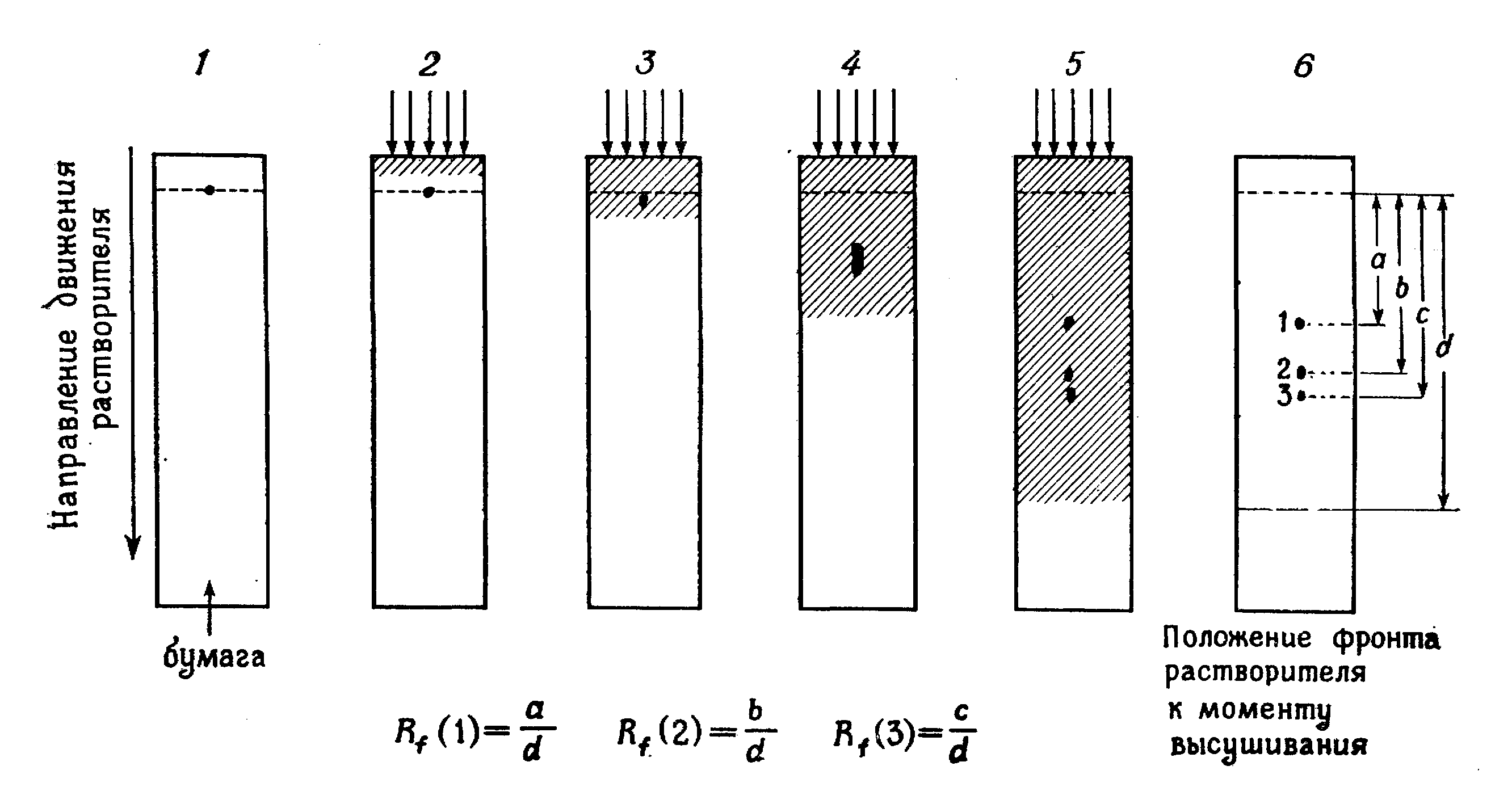
Електрофорез сироватки крові проводять в 1% агарі в мединал-вероналовому буфері (рН 8,6) з іонною силою 0,05. Усі білки сироватки при рН 8,6 заряджені негативно і переміщаються в електричному полі в напрямі до анода. Однак практично всі білкові фракції сироватки, які рухаються повільно, переміщаються до катода. Аномальний напрям руху цих білків пояснюєюь наявністю в агаровому гелі електроендоосмотичного струму рідини, спрямованого від анода до катода, а також струму рідини, який виникає під час нерівномірного випаровування води з поверхні гелю. Випаровування призводить до нерівномірного розподілу електричного поля в гелі. Зменшити випаровування води можна трьома способами:
1) герметично закривати електрофоретичну камеру під час електрофорезу;
2) проводити електрофорез за низьких температур у холодильнику або в холодній кімнаті;
3) буферний розчин повинен бути попередньо охолоджений.
Реактиви
1. Веронал-мединаловий буфер (рН 8,6; іонна сила 0,1): у 500 мл дистильованої води розчиняють 17,52 г мединалу, додають 2,76 г вероналу і, переміщуючи, нагрівають у теплій водяній бані до розчинення вероналу. Тоді об’єм розчину доводять дистильованою водою до 1 л.
2. Агар – 1% розчин. Агар повинен бути чистим і прозорим. Готують 2% розчин на дистильованій воді. Гарячий розчин розливають у ванночки товщиною 1,0–1,5 см. Після застигання агарову пластинку розрізають на кубики, переносять їх у товстостінний стакан і промивають протягом двох діб під проточною водопровідною водою, тоді протягом доби – дистильованою водою, змінюючи її чотири–п’ять разів. Агар підсушують фільтрувальним папером, переносять у колбу, розігрівають, гарячий розчин розводять у два рази буферним розчином і тричі фільтрують через три шари марлі. Розчин арагу зберігають у холодильнику. Перед нанесенням на пластинку його нагрівають на киплячій водяній бані доти, поки в агарі не зникнуть пухирці повітря.
3. Амідовий чорний 10Б – 0,1% розчин у 2% оцтовій кислоті.
4. СН3СООН – 5% розчин.
Приготування агарових пластинок і проведення електрофорезу. Для приготування агарових пластинок використовують предметні скельця. Попередньо їх знежирюють, ретельно промивають водою і висушують. Скельця нумерують, викладають на строго горизонтальну поверхню і на кожне скельце обережно наливають піпеткою із широким носиком 4 мл агару. Пухирці повітря в агарі необхідно вивести піпеткою на край скла. Коли агар застигне і утвориться твердий гель, у ньому посередині вирізають лунку (кишеньку) для внесення досліджуваного розчину. Підготовлені скельця складають в електрофоретичну камеру. Агарове плато з’єднують з буферним розчином містками з фільтрувального паперу. В кожну лунку на пластинці вносять сироватку крові, попередньо розведену буферним розчином. На електрофореграму наносять приблизно по 100–300 мкг білка в об’ємі 0,01 мл. Прилад підключають до джерела струму. Повільно підвищуючи напругу, доводять силу струму до 20–25 мА (за напруги 200–300 В), електрофорез проводять протягом 3–4 год.
Обробка агарових пластинок. Після завершення електрофорезу струм вимикають, предметні скельця з агаровим гелем відразу виймають з камери і поміщають на 30 хв у 5% розчин СН3СООН. Кожну пластинку покривають смужкою фільтрувального паперу, змоченого в 5% розчині СН3СООН, для видалення води та солей, а згодом висушують за кімнатної температури протягом ночі. Для пришвидшення процесу висушування його проводять при 37°С або під потоком теплого повітря. Папір, який може приклеїтися до агарового шару, обережно знімають, попередньо розмочивши його водою. Перед зафарбовуванням агарове поле повинне бути абсолютно сухим і прозорим. Зафарбовування зразків на електрофореграмах проводять амідовим чорним 10Б протягом ночі. Фарбу, яка не зв’язалася з білками, відмивають 5% розчином СН3СООН. Відмиті електрофореграми висушують при 37°С.
Концентрувальний та розділювальний поліакриламідні гелі. Концентрація поліакриламіду в гелі може бути однорідною або градієнтною. Найчастіше концентрація поліакриламіду становить 10%, що дає змогу розділяти білки з молекулярною масою 10–150 кДа. Якщо необхідно аналізувати невідомі білки або використати більш широкий діапазон розділення білків, то застосовують градієнтні гелі. Наприклад, 4–12% трис-гліциновий гель є зручним для розділення білків у діапазоні від 30 до 200 кДа, тоді як 1–20% гелі зручні для розділення білків від 6 до 150 кДа. Зазвичай товщина SDS-ПААГ гелів становить 1,0 або 1,5 мм; проте для блотингу найліпше використовувати гелі завтовшки 1 мм. Компоненти, необхідні для приготування поліакриламідних гелів, змішують у послідовності (!) та співвідношеннях, неведених у таблиці.
Таблиця
Розділювальний поліакриламідний гель |
8% |
10% |
11% |
12% |
15% |
17,5% |
20% |
|
15 мл |
H2O (мл) |
6,95 |
5,95 |
5,45 |
4,95 |
3,45 |
2,2 |
0,95 |
30% акриламідна суміш (мл) |
4,0 |
5,0 |
5,5 |
6,0 |
7,5 |
8,75 |
10,0 |
|
1,5 М трис-HCl, pH 8,8 (мл) |
3,75 |
3,75 |
3,75 |
3,75 |
3,75 |
3,75 |
3,75 |
|
10% SDS (мл) |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
|
10% персульфат амонію (APS) (мл) |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
0,15 |
|
TEMED* (мл) |
0,006 |
0,006 |
0,006 |
0,006 |
0,006 |
0,006 |
0,006 |
|
Концентруючий поліакриламідний гель |
5% |
|
||||||
4 мл |
H2O (мл) |
2,76 |
||||||
30% акриламідна суміш (мл) |
0,66 |
|||||||
1,0 М трис-HCl, pH 6,8 (мл) |
0,5 |
|||||||
10% SDS (мл) |
0,04 |
|||||||
10% персульфат амонію (мл) |
0,04 |
|||||||
TEMED* (мл) |
0,004 |
|||||||
* N,N,N,N-tetrametyhyl-Ethylenediamin (N,N,N,N-тетраметил-етилендіамін)
Суміш компонентів розділювального поліакриламідного гелю обережно вносять у простір між двома скляними пластинками, після чого нашаровують 0,1% SDS для 10% гелів і менше або ізобутанол для гелів більшої концентрації. Через 30 хв (після полімеризації гелю) нашарований SDS або ізобутанол видаляють за допомогою фільтрувального паперу, а між пластинками заливають концентрувальний гель і вставляють гребінку. Після полімеризації концентрувального гелю, яка відбувається протягом 40 хв, скляні пластинки з гелем поміщають у камеру для електрофорезу.
Електродні буфери. Для проведення електрофорезу білків найчастіше використовують два типи буферів: трис-гліциновий і трис-трициновий. Ці електродні буфери можуть включати 0,1% детергент, найчастіше SDS. Система трис-гліцинового буфера є зручною для розділення білків у широкому діапазоні молекулярних мас (6–200 кДа), її можна використовувати для електрофорезів білків як у денатуруючих умовах, так і в неденатуруючих. Трис-тріцинова система є ліпшою для розділення білків з низькою молекулярною масою (<10 кДа), які потребують попередньої денатурації та відновлення перед нанесенням у гель. Обидві буферні системи сумісні з перенесенням білків на мікропорову мембрану. Зазвичай електродні буфери готують 10-кратної концентрації і розводять безпосередньо перед використанням.
Десятикратний трис-гліциновий буфер, рН 8,3
1. Трис – 25 мM розчин.
2. Гліцин – 192 мM розчин.
3. SDS – 0,1% розчин.
Десятикратний трис-тріциновий буфер, рН 8,3
1. Трис – 100 мM розчин.
2. Тріцин – 100 мM розчин.
3. SDS – 0,1% розчин.
Електродний буфер заливають у камеру для електрофорезу білків в об’ємі, який забезпечує повне занурення лунок концентрувального гелю, після чого в комірки концентрувального гелю вносять зразки. Електрофорез проводять за сили струму 20 мА, напруги 200 В та потужності 50 Вт на пластинку розміром 1015 см.




Повтор
Компактність білкової зони може залежати від способу нанесення зразка (див. нижче). Настільки ж важливо звернути увагу на поведінку виходить з колонки білкової зони. Перемішування білкових зон в поздовжньому напрямку при використанні занадто широких колонок - часта помилка, BO-БАГАТЬОХ випадках зводить нанівець результати чудової-поділу. З тієї ж причини проточні кювети для реєстрації кількості білка повинні мати малі розміри, щоб звести до мінімуму можливість такого перемішування; крім того, їх слід поміщати якомога ближче до вихідного отвору колонки (рис. 5.10).
Колонку необхідно заповнювати по можливості в один прийом, щоб запобігти поділ гранул за розмірами »процесі осідання (див. рис. 1.11). З цією метою використовують суспензію, обсяг якої не більш ніж в два рази перевищує обсяг осів матеріалу. Заповнення колонки слід проводити якомога швидше без використання тиску. Хороший спосіб створити більш високу швидкість потоку - це приєднати знизу до колонки довгу, але не дуже вузьку трубку, з тим щоб збільшити загальну висоту стовпа рідини; тоді падіння тиску в самій колонці виявиться вище.
Дуже важливою умовою гарного поділу є нанесення зразка. Чим рівніше він нанесений, тим краще буде кінцевий результат. З успіхом використовуються аплікатори, завдяки вдалій конструкції яких можна домогтися рівномірного потоку рідини через всю поверхню гелю (див. рис. 1.8). Не можна допускати, щоб між поверхнею гелю і аплікатором створювалося значне мертвий простір, так як це призводить до змішування шарів (див. рис. 1.10). Не слід застосовувати розчини зразка, мають меншу щільність, ніж буфер (наприклад, розчини, що містять ацетон), оскільки при подальшому промиванні колонки цим буфером він буде прагнути витіснити вгору останні порції зразка і ці порції будуть повільно просочуватися в гель вже після того, як весь решті зразок увійде в гель. Аплікатор слід спробувати з допомогою пофарбованого зразка: якщо зона ■ зразка буде нерівномірною, краще нанести його вручну. Для цього аплікатор видаляють, дають буферу вбратися в поверхню гелю, закривають затискач на вихідний трубці і потім акуратно наносять зразок за допомогою пастерівською піпетки, даючи йому проникнути на 5-10 мм всередину шару гелю, і в той же час переміщують піпетку по периметру колонки ( рис. 5.11).
Рис. 5.10. Неправильний (А) і правильний (Б) способи установки колонки для гель-фільтрації (або інший) по відношенню до УФ-монітора і ■ колектору фракцій. Довжина сполучної трубки повинна бути мінімальною, причому трубка повинна бути як можна вже, але не ускладнювати потоку.
Рис. 5.11. Нанесення зразка на поверхню гель-фільтрувальної колонки вручну. Слід дотримуватися обережності, щоб не порушити рівну поверхню гелю. А. Буфер дають повністю вбратися в гель, після чого закривають кран. Б. Зразок наносять на колонку за допомогою піпетки; відкривають кран. В. Дають зразком проникнути в гель. Г. Зразок повністю вбрався в гель; закривають кран. Д. Наносять буфер і відкривають кран. Є. Дають буферу повністю вбратися в гель і закривають кран. Ж. Наносять ще якийсь обсяг буфера і приєднують до колонки аплікатор; мертвий простір у верхній частині колонки допустимо.
Коли весь зразок буде завдано, зажим відкривають і дають зразком вбратися в поверхню гелю. Потім повторюють весь процес, використовуючи буфер (приблизно той же об'єм, що і обсяг зразка), - дають йому ввійти в гель, додають ще трохи буфера і приєднують аплікатор для безперервної подачі буфера.
Слід пам'ятати, що фермент буде елюіроваться з колонки в тому буфері, який міститься в ній до початку гель-фільтрації. Тому неважливо, в якому буфері зразок наноситься на колонку або елюіруется з неї. Таким чином, якщо потрібний фермент необхідно елюіровать в якомусь особливому і щодо дорогому буфері, немає сенсу витрачати його даремно. Перед тим як нанести зразок, потрібно промити колонку об'ємом цього спеціального буфера, рівним одному обсягом колонки. Потім проводять гель-фільтрацію, використовуючи будь-якої звичайний дешевий буферний розчин.
Гравітаційна нестійкість, яка виникає в ході нанесення зразка (див. рис. 5.4), особливо велика, коли використовуються дуже щільні розчини, такі, як розчинені опади, отримані при фракціонуванні сульфатом амонію. Це особливо важка проблема в разі застосування м'яких гелів "типу сефадексе G-200. При використанні сучасних матеріалів з жорсткими зернами щільна поверхня хроматографії-чеського шару дозволяє запобігти надмірне змішування при нашарування щільного розчину на менш щільний буфер. Проте більш високий дозвіл (за рахунок того, що зона поділюваних речовин утворює дуже тонкий і щільний шар) виходить, якщо зразок наносять знизу колонки при висхідному потоці рідини, а потім перевертають колонку так, що менш щільні шари буфера опиняються під поверхнею гелю (рис. 5.12). Це не завжди зручно робити, але варто спробувати, «їли дозвіл гірше, ніж очікувалося.
Рис. 5.12. Схема, що демонструє, яким чином можна уникнути гравітаційної нестійкості, що на рис. 5.5. А. Щільний розчин про-• зразка наносять знизу колонки при висхідному потоці рідини. Б. Після того як весь зразок завдано, колонку перевертають. В. Менш щільний Йуфер слід за зразком - колонка промивається низхідним потоком.
Нижче підсумовані практичні рекомендації. Розміри "колонки повинні в 30-100 разів перевищувати обсяг зразка. Вихідна концентрація білка в ідеалі повинна становити 10 - '20 мг-мл" 1, найбільше - 30 мг-мл "1. Таким чином, 100 мг білка слід розчинити в обсязі не меншому ніж 3,5 мл, краще 5-6 мл, і використовувати колонку об'ємом 200-250 см3. Довжина колонки повинна в 20-40 разів перевищувати її діаметр, тобто для вказаної кількості білка підходящими будуть колонки діаметром 2,5 і довжиною 50 см або відповідно 1,8 і 100 см. Ідеальні розміри колонки можна визначити наступним чином: діаметр = ут/10 см, де т - кількість білка в мг, а довжина = 30Хдіаметр.
Швидкості потоку обмежуються максимально досяжними швидкостями протікання буфера. Сучасні жорсткі мате-
ріали витримують швидкість потоку, що перевищує швидкість встановлення рівноваги, тому необхідно знати рекомендації фірм-виробників. Для сефакрілов придатні швидкості потоку до 30 см-ч "1 (30 мл-ч ^-см" 2), але для ультрагелей рекомендуються менші швидкості (див. також гл. 7). Час поділу можна обчислити, знаючи розміри колонки. Якщо фермент виходить в середині інтервалу фракціонування для сефакрілов, то в наведеному вище прикладі з колонкою діаметром 2,5 і довжиною 50 см він буде з'являтися в елюатів приблизно через 2 години після нанесення (при швидкості потоку 30 см-ч ^ 1). Для колонки розміром 1,8 x100 см часом буде в два рази більше, хоча на довгій колонці дозвіл може бути декілька вище. Як вже зазначалося,
більш жорсткі матеріали з дрібними гранулами дозволяють швидше розділяти речовини під тиском, і цілком імовірно, що методи із застосуванням помірного тиску (проміжні між рутинними операціями, проведеними при низькому тиску, і швидкими операціями під високим тиском з використанням техніки ЖХВД) отримають більш широке поширення.