

Этап 4 Скрининг новорожденных, исследование кк.
В четвертой и заключительной фазе этого исследования, чтобы увеличить размер выборки для дальнейшей проверки 2-х уровневнего подхода к идентификации МДД в период новорожденности, мы взяли образцы у большой когорты новорожденных анонимно. Это увеличило наши выборки на 19 884 новорожденных мальчиков (общее число 37 649). На основании результатов фазы 2 и фазы 3 исследования, мы ограничили скрининг ДНК на сухой капле крови, чтобы мальчики с КК ≥ 750U / л. Существовали 308 уровней КФК оказались > 750U / л, и десять > 2000 Ед / л. В этом заключительном этапе исследования мы также включили анализ КК сухих пятен крови новорожденных девочек 18 763. Для женщин, КK был ≥ 750U / л в 242, и КK ≥ 2000 2 анонимных сухих пятен крови.
Анализ днк сухой капли крови
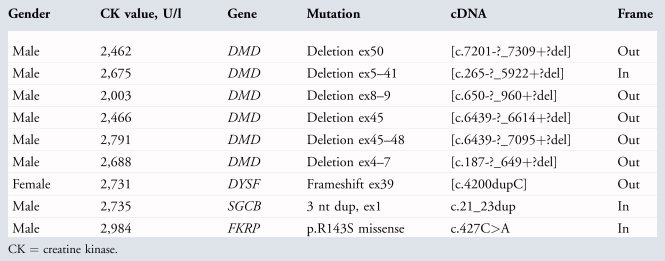
Среди 37 649 новорожденных обследованых на МДД (2 фаза, 3, 4), у 6 мальчиков были обнаружены мутации в гене дистрофина. Все они были делециями одного или нескольких экзонов, 5 с нарушением рамки считывания и 1 без нарушения рамки считывания (табл. 2), ни точечные мутаций или дупликаций обнаружено не было. Эти мутации следуют типичному распределению наблюдаемому в большой когорте, хотя о делеции с сохранением рамки считывания экзонов 5-41 сообщалось прежде только один раз (http://www.leiden.nl), и было связано с МДД, вероятно, в связи с делецией, которая охватывает критическую актин-связывающую область.
Дискуссия.
КK тестирование сухой капли крови для выявления случаев МДД в период новорожденности было утверждено в 1979 году и рассчитано на активность фермента катализировать трансфосфорилирование АДФ в ATФ. Как первоначально введено, на основе анализа биолюминесценции установлена интенсивность активности фермента; последующие изменения использованного НАД Н на основе флуорометрического считывания как меры активности КK. Таблица 3 отслеживает последовательные истории NBS для выявления МДД я в Новой-Зеландии 1 через программы в Эдинбурге, 2 Германии, 3 Канаде, 4 Франции, 5 США (западная Пенсильвания), 6 Уэльсе, Кипре 7, 8. Антверпен является единственной программой, которая поддерживает NBS для МДД и по сей день. В рамках этой программы, образцы с повышенным КK, повторно исследованы в венозной крови, взятой примерно в 6 недель после родов.
С самого начала наши цели включали создание программы NBS, которая будет соответствовать практике акушерства в США, где мать и ребенок выписывается в течение 24 -48 часов после неосложненных родов, и разработка метода должна включать возможность легко отличать ложные и истинные результаты. Выполнение этой задачи требует 2-х уровневой системы анализа: тестирования КK, за которым следуют анализ ДНК тех же сухих пятен крови. Система имеет сходство с программой NBS для муковисцидоза на основе 2-х уровневого молекулярного генетического тестирования, которая была впервые представлена в рамках пилотной программы Wisconsin. Утвержденный метод был необходим для извлечения геномной ДНК из небольшого сухого пятна крови, после чего весь геном исследуется на наличие делеци / дупликаций в гене дистрофина с использованием SCAIP в сочетании с MLPA. Подготовительные исследования при условии уверенности в методологии, основанной на 100% точности в выявлении делеций 7 экзона и дупликацию 6 экзона взятых у пациентов с МДД с известными мутациями, размещенные на карты обследования новорожденных в больнице и отправлен в клиническую ДНК лабораторию в Университете штата Юта. Кроме того, необходимо создать уровни диапазона КК для анонимных сухие пятна крови. Это важное мероприятие стало возможным благодаря всестороннему сотрудничеству лаборатории ODH. По анонимным анализам КК > 30 000 новорожденных (см. рис 1), мы установили, отправную точку для анализа ДНК по уровню КK на 3 стандартных отклонения выше среднего. Добавление КK тестирования к набору тестов сухой капли крови ODH было не слишком обременительной, а стоимость добавления этого 1 анализа (до 35 других) была минимальной (около $ 1,00 ). Для превышающих порог КK требуется анализ ДНК, стоимость в лаборатории Университета штата Юта дополнительно $ 150.00 .
Результаты нашего исследования подтверждают, что 2-х уровневая система анализа для скрининга новорожденных на МДД, возможно, принесет еще большее удовлетворение, чем ожидалось. В ходе этой программы, мы обследование 37 649 мальчиков и обнаружили 6 с мутациями гена дистрофина, частота 1 к 6291. Сравнительная заболеваемость новорожденных мальчиков МДД документально ниже, чем в других исследованиях по всему миру, которые варьировались от 1 к 3802 до 1 к 6002 (с учетом всех программ вместе, 1 к 4087, см. таблицу 3), и должны рассматриваться с осторожностью в зависимости от размера выборки и места в государстве в США. Что особенно заметно по нашему исследованию, что все наши пациенты с МДД (или дистрофинопатией) имели КФК при рождении ≥ 2000 Ед / л. Это разница между документально подтвержденными случаями МДД и с повышенными КK, у которых не обнаружены мутации, обеспечивает достаточную уверенность для ложных результатов, что позволяет нам повысить порог для анализа ДНК в фазе 3 исследование КК ≥ 750U / л. Это привело к снижению числа новорожденных, которым необходимо тестирование на мутации, примерно 68%, что представляет собой значительную экономию средств по данной программе . С дополнительным подтверждением наших выводов, эти первоначальные исследования показывают, что порог для анализа ДНК может быть поднят еще выше (например, КK ≥ 1000 Ед / л), повышение потенциального соотношения затрат и выгоды.
По мере того как развивается наша программа, у нас было больше уверенности в идентификации большего количества мутаций МДД, но мы знали об ограничениях. Дополнительный тест должен будет подтвердить, что точечные мутации, присутствующие примерно у четверти пациентов с МДД, не обнаруженые в этом исследовании, обнаружены с использованием проверки ДНК, выделенной из сухих пятен крови. Тем не менее, мы уверены, что соответствующая методология была применена в данном исследовании, на основе наших предыдущих работ демонстрирующих обнаружение 506 точечных мутаций (294 нонсенс мутаций) при анализе 1111 дистрофинопатий представляющих 46% пациентов (более широко представлены в этой группе населения из-за дизайна исследования). Кроме того, группа дистрофинопатий проявляется преимущественно как кардиомиопатия, щадящая скелетные мышцы (например, Х-сцепленных кардиомиопатия) часто не входит в протокол NBS.
Хорошо известно, что многие пациенты в этой группе имели уменьшение КФК в венозной крови, а некоторые даже нормальный или почти нормальный уровень, мы также выявили новорожденных на другом конце спектра с повышенным КK и но не имевших МДД. По этой причине, мы расширили исследование для решения этой задачи. На заключительном этапе этого исследования, мы сделали анализ ДНК для наиболее распространенных генов конечностных миодистрофий, если КK был ≥ 2000 Ед / л при отсутствии определеных мутаций гена дистрофина. В этом небольшом примере мы обнаружили, 1 человека с известной однонуклеотидной мутацией в DYSF, 1 - с известной миссенс мутацией в FKRP, и еще с дупликацией SGCB неизвестной патогенности, которые были зарегистрированы у 5 пациентов (Лейден, см. таблицу 2). Эти данные показывают доказательство принципа, что генные мутации КПМД могут быть определены как часть процесса отбора. Только 1 патогенный аллель был обнаружен в каждом конкретном случае, что не редкость для этих генов. Дальнейшая характеристика этих образцов должна будет оценить число изменений, указывающих на вторую, незамеченную большую делецию или дупликацию.
Наше завершенное исследование не было предназначено для решения вопроса о том должны ли быть введены эти тесты, чтобы обеспечить путь для реализации терапевтической пользы для МДД. Фаза 2 программы изучила этические вопросы, связанные с путем оценки родителей через анкеты, однако, эта тема была зарезервирована для будущей статьи. Программа, которую мы ввели отличается от прошлых программ и нынешний подход для МДД требует 3 этапа: (1) КK тестирование сухой капли крови, а затем (2) подтверждение повышенного уровня КФК при анализе венозной крови, полученной на 4 - 6 недели, (3) последний шаг, который требует дополнительного анализа кровь для анализа ДНК. Подход, который мы разработали в 2 этапа, со всеми испытаниями, проведенными с использованием крови, полученной из пятки в течение первых 24 - 48 часов. Все тестирование проводилось из того же сухого пятна крови. Пороговый уровень КК определяет, будет ли проведено ДНК тестирование без дополнительного анализа крови, полученной от новорожденных. ДНК тест использует самые современные технологии available40. Лечение развивается настолько далеко, что оправдан скрининг для новорожденных на МДД требует оценки государственных и федеральных агентств с соответствующей юрисдикцией. Если будет разработана ранняя терапия, что повысит уровень здоровья для людей с МДД, наше исследование служит в качестве модели для осуществления скрининга новорожденных на МДД. Если разработка перспективной терапии МДД продолжит идти нынешними темпами, скрининг новорожденных может быть на страже этой болезни, не только в США, но и в других странах. В случае успеха терапии дистрофинопатий доступной для новорожденных, принципы исследования должны быть установлены.
Ссылки
1 Drummond LM. Creatine phosphokinase levels in the newborn and their use in screening for Duchenne muscular dystrophy. Arch Dis Child 1979; 54: 362–366.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 15
2 Skinner R, Emery AEH, Scheuerbrandt G, Syme J. Feasibility of neonatal screening for Duchenne muscular dystrophy. J Med Genet 1982; 19: 1–3.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 12
3 Scheuerbrandt G, Lövgren T, Mortier W. Screening for Duchenne muscular dystrophy: an improved screening test for creatine kinase and its application in an infant screening program. Muscle Nerve 1986; 9: 11–23.
Direct Link: AbstractPDF(1108K)References
4 Greenberg CR, Jacobs HK, Nylen E, et al. Gene studies in newborn males with Duchenne muscular dystrophy detected by neonatal screening. Lancet 1988; 2: 425–427.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 14
5 Plauchu H, Dorche C, Cordier MP, et al. Duchenne muscular dystrophy: neonatal screening and prenatal diagnosis. Lancet 1989; 1: 669.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 4
6 Naylor EW. New technologies in newborn screening. Yale J Biol Med 1991; 64: 21–24.
PubMed,CAS,Web of Science® Times Cited: 3
7 Bradley DM, Parsons EP, Clarke AJ. Experience with screening newborns for Duchenne muscular dystrophy in Wales. BMJ 1993; 306: 357–360.
CrossRef,PubMed,CAS
8 Drousiotou A, Ioannou P, Georgiou T, et al. Neonatal screening for Duchenne muscular dystrophy: a novel semiquantitative application of bioluminescence test for creatine kinase in a pilot national program in Cyprus. Genet Test 1998; 2: 55–60.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 17
9 Eyskens F, Philips E. Newborn screening for Duchenne muscular dystrophy. The experience in the province of Antwerp. Neuromuscul Disord 2006; 16: 721.
CrossRef,Web of Science®
10 Pearce JM, Pennington RJ, Walton JN. Serum enzyme studies in muscle disease. III. Serum creatine kinase activity in relatives of patients with Duchenne type muscular dystrophy. J Neurol Neurosurg Psychiatry 1964; 27: 181–185.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 95
11 Heyck H, Laudahn G, Carsten P. Enzyme activity determination in progressive muscular dystrophy. IV. Serum enzymatic kinetics in the preclinical stage of the Duchenne type during the 1st 2 years of life [in German]. Klin Wochenschr 1966; 44: 695–700.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 26
12 Zellweger H, Antonik A. Newborn screening for Duchenne muscular dystrophy. Pediatrics 1975; 55: 30–34.
PubMed,CAS,Web of Science® Times Cited: 83
13 Wilson JMG, Jungner G. Principles and practice of screening for disease. Public Health Paper No. 34. Geneva, Switzerland: World Health Organization, 1968.
14 Ross LF. Screening for conditions that do not meet the Wilson and Jungner criteria: the case of Duchenne muscular dystrophy. Am J Med Genet A 2006; 140: 914–922. PubMed
15 Goemans NM, Tulinius M, van den Akker JT, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med 2011; 364: 1513–1522.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 26
16 Kinali M, Arechavala-Gomeza V, Feng L, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo controlled, dose-escalation, proof-of-concept study. Lancet Neurol 2009; 8: 918–928.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 128
17 Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in Duchenne muscular dystrophy patients after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011; 378: 595–605.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 11
18 Malik V, Rodino-Klapac LR, Viollet L, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol 2010; 67: 771–780.
PubMed,CAS,Web of Science® Times Cited: 26
19 Finkel R. Read-through strategies for suppression of nonsense mutations in Duchenne/Becker muscular dystrophy: aminoglycosides and Ataluren (PTC124). J Child Neurol 2010; 25: 1158–1164.
CrossRef,PubMed,Web of Science® Times Cited: 11
20 Moxley RT III, Pandya S. Weekend high-dose prednisone: a new option for treatment of Duchenne muscular dystrophy. Neurology 2011; 77: 416–417.
CrossRef,PubMed,Web of Science®
21 Balaban B, Matthews DJ, Clayton GH, Carry T. Corticosteroid treatment and functional improvement in Duchenne muscular dystrophy: long-term effect. Am J Phys Med Rehabil 2005; 84: 843–850.
CrossRef,PubMed,Web of Science® Times Cited: 38
22 Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord 2006; 16: 249–255.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 55
23 King WM, Ruttencutter R, Nagaraja HN, et al. Orthopedic outcomes of long-term daily corticosteroid treatment in Duchenne muscular dystrophy. Neurology 2007; 68: 1607–1613.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 31
24 Houde S, Filiatrault M, Fournier A, et al. Deflazacort use in Duchenne muscular dystrophy: an 8-year follow-up. Pediatr Neurol 2008; 38: 200–206.
CrossRef,PubMed,Web of Science® Times Cited: 19
25 Moxley RT III, Pandya S, Ciafaloni E, et al. Change in natural history of Duchenne muscular dystrophy with long-term corticosteroid treatment: implications for management. J Child Neurol 2010; 25: 1116–1129.
CrossRef,PubMed,Web of Science® Times Cited: 4
26 Moxley RT III, Ashwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005; 64: 13–20.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 74
27 Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev 2008;( 1): CD003725.
PubMed,CAS
28 Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010; 9: 77–93.
CrossRef,PubMed,Web of Science® Times Cited: 68
29 Ciafaloni E, Fox DJ, Pandya S, et al. Delayed diagnosis in Duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research network (MD STARnet). J Pediatr 2009; 155: 380–385.
CrossRef,PubMed,Web of Science® Times Cited: 13
30 Rosalki SB. An improved procedure for serum creatine phosphokinase determination. J Lab Clin Med 1967; 69: 696–705.
PubMed,CAS,Web of Science® Times Cited: 1661
31 Orfanos AP, Naylor EW. A rapid screening test for Duchenne muscular dystrophy using dried blood spot specimens. Clin Chim Acta 1984; 138: 267–274.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 11
32 Flanigan KM, von Niederhausern A, Dunn DM, et al. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet 2003; 72: 931–939.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 77
33 Lalic T, Vossen RH, Coffa J, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet 2005; 13: 1231–1234.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 45
34сAhn JW, Ogilvie CM, Welch A, et al. Detection of subtelomere imbalance using MLPA: validation, development of an analysis protocol, and application in a diagnostic centre. BMC Med Genet 2007; 8: 9.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 12
35 Rudolph N, Gross RT. Creatine kinase activity in serum of newborn infants as indicator of fetal trauma during birth. Pediatrics 1966; 38: 1039–1046.
PubMed,CAS,Web of Science® Times Cited: 36
36 Bodensteiner JB, Zellweger H. Creatine phosphokinase in normal neonates and young infants. J Lab Clin Med 1971; 77: 853–858.
PubMed,CAS,Web of Science® Times Cited: 40
37 Gilboa N, Swanson JR. Serum creatine phosphokinase in normal newborns. Arch Dis Child 1976; 51: 283–285.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 32
38 Gregg, RG, Simantel A, Farrell PM, et al. Newborn screening for cystic fibrosis in Wisconsin: comparison of biochemical and molecular methods. Pedatrics 1997; 99: 819–824.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 75
39 van Ommen GJB, Scheuerbrandt G. Neonatal screening for muscular dystrophy. Consensus recommendation of the 14th workshop sponsored by the European Neuromuscular Center (ENMC). Neuromuscul Disord 1993; 3: 231–239.
CrossRef,PubMed,CAS
40 Flanigan KM, Dunn DM, von Niederhausern A, et al. Mutational spectrum of DMD mutations in dystrophinopathy patients: application of modern diagnostic techniques to a large cohort. Hum Mutat 2009; 30: 1657–1666.
Direct Link: AbstractPDF(316K)References
41 Arbustini E, Diegoli M, Morbini P, et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol 2000; 35: 1760–1768.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 31
42 Kimura S, Ikezawa M, Ozasa S, et al. Novel mutation in splicing donor of dystrophin gene first exon in a patient with dilated cardiomyopathy but no clinical signs of skeletal myopathy. J Child Neurol 2007; 22: 901–906.
CrossRef,PubMed,Web of Science® Times Cited: 4
43 Feng J, Yan J, Buzin CH, et al. Mutations in the dystrophin gene are associated with sporadic dilated cardiomyopathy. Mol Genet Metab 2002; 77: 119–126.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 32
44 Nguyen K, Bassez G, Krahn M, et al. Phenotypic study in 40 patients with dysferlin gene mutations: high frequency of atypical phenotypes. Arch Neurol 2007; 64: 1176–1182.
CrossRef,PubMed,Web of Science® Times Cited: 50
45 Trabelsi M, Kavian N, Daoud F, et al. Revised spectrum of mutations in sarcoglycanopathies. Eur J Hum Genet 2008; 16: 793–803.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 9
46 Brockington M, Yuva Y, Prandi P, et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum Mol Genet 2001; 10: 2851–2859.
CrossRef,PubMed,CAS,Web of Science® Times Cited: 215
Оригинальна статья : http://onlinelibrary.wiley.com/doi/10.1002/ana.23528/full
Переведено проектом МОЙМИО: http://www.mymio.org/