

КОЛЕБАТЕЛЬНЫЕ
РЕАКЦИИ,
р-ции, в ходе к-рых концентрации
промежут. соединений и скорость р-ции
испытывают колебания. Колебания м. б.
периодическими, в этом случае значения
c(t) колеблющихся концентраций
(t - время) можно представить рядом Фурье:
![]() где
аn,
bn
- коэффициенты разложения ф-ции c(t) в рад
(амплитуды отдельных гармонич. компонент),
An
-
комплексные амплитуды, -
частота колебаний (i - мнимая единица).
В общем случае амплитуды и частоты
колебаний могут изменяться во времени
(колебания затухающие, нарастающие,
модулированные). Колебания концентраций
промежут. соед. могут быть непериодическими
или иметь непрерывный спектр.
Колебания
концентраций
промежут. соед. - относительно редкое
явление, наблюдаемое в ходе нек-рых
сложных р-ций. Элементарные хим. р-ции
являются релаксац. процессами,
обеспечивающими монотонное приближение
реагирующей системы к состоянию
термодинамич. равновесия.
Для возникновения колебаний в ходе
гомог. изотермич. р-ции необходимо
наличие промежут. соед. и взаимодействие
между ними. В открытых
системах
существуют стационарные состояния, в
к-рых концентрация
c(i) i-го промежут. соед. не зависит от
времени (сi=c0i).
При небольших отклонениях системы от
стационарного состояния изменение сi
описывается суммой экспонент с
комплексными показателями:
где
аn,
bn
- коэффициенты разложения ф-ции c(t) в рад
(амплитуды отдельных гармонич. компонент),
An
-
комплексные амплитуды, -
частота колебаний (i - мнимая единица).
В общем случае амплитуды и частоты
колебаний могут изменяться во времени
(колебания затухающие, нарастающие,
модулированные). Колебания концентраций
промежут. соед. могут быть непериодическими
или иметь непрерывный спектр.
Колебания
концентраций
промежут. соед. - относительно редкое
явление, наблюдаемое в ходе нек-рых
сложных р-ций. Элементарные хим. р-ции
являются релаксац. процессами,
обеспечивающими монотонное приближение
реагирующей системы к состоянию
термодинамич. равновесия.
Для возникновения колебаний в ходе
гомог. изотермич. р-ции необходимо
наличие промежут. соед. и взаимодействие
между ними. В открытых
системах
существуют стационарные состояния, в
к-рых концентрация
c(i) i-го промежут. соед. не зависит от
времени (сi=c0i).
При небольших отклонениях системы от
стационарного состояния изменение сi
описывается суммой экспонент с
комплексными показателями:
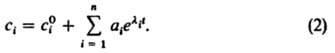 Величины
i=i+ii,
наз. характеристич. числами. В неколебат.
устойчивых системах i
отрицательны и действительны (i<0,
i=0).
В этих случаях обычно вместо i
используют времена релаксации
i=1/i.
Если стационарное состояние достаточно
близко к состоянию термодинамич.
равновесия
(выполняются соотношения взаимности
Онсагера, см. Термодинамика
необратимых процессов),
то все i
действительны и отрицательны (теорема
Пригожина).
В этом случае система приближается к
стационарному состоянию без колебаний.
В сильно неравновесных системах i
могут стать комплексными числами, что
соответствует появлению колебаний
около стационарного состояния. При
определенных значениях параметров
сильно неравновесной системы (концентраций
исходных реагентов,
т-ры, давления
и т.д.) стационарное состояние может
потерять устойчивость. Потеря устойчивости
стационарного состояния является
частным случаем бифуркации, т.е. изменения
при определенном (бифуркационном)
значении к.-л. параметра числа или типа
разл. кинетич. режимов системы. Имеется
два простейших случая бифуркации
устойчивого стационарного состояния.
В первом случае одно i
становится положительным. При этом в
точке бифуркации (i=0)
исходно устойчивое состояние становится
неустойчивым или сливается с неустойчивым
стационарным состоянием и исчезает, а
система переходит в новое устойчивое
состояние. В пространстве параметров
в окрестности этой бифуркации существует
область, где система обладает по крайней
мере тремя стационарными состояниями,
из к-рых два устойчивы, а одно неустойчиво.
Во втором случае действит. часть одной
пары
комплексных характеристич. чисел
становится положительной. При этом в
окрестности потерявшего устойчивость
стационарного состояния возникают
устойчивые колебания. После прохождения
точки бифуркации при дальнейшем изменении
параметра количеств, характеристики
колебаний (частота, амплитуда и т.д.)
могут сильно меняться, но качеств. тип
поведения системы сохраняется. В хим.
системах неустойчивости могут возникать
в результате ускорения р-ции ее продуктами
или др. видов автокатализа,
субстратного или перекрестного
ингибирования (см. Ингибиторы),
конкуренции исходных в-в за промежут.
соед. и т.п. В неизотермич. системах
причиной неустойчивости может служить
самоускорение экзотермич. стадий р-ции,
а в электрохим. р-циях экспоненциальная
зависимость скорости р-ции от поляризации
электродов.
Появление простейших неустойчивостей
и соответствующих кинетич. состояний
системы удобно пояснить на примере
ферментативной р-ции с двумя
субстратами
S1
и S2,
один из к-рых, напр. S1,
ингибирует фермент
Е:
S01S1
S02S2
S1+E1
S1E
S1E+S2S1E:P
S1E+S1S1S1E
Субстраты
S1
и S2
могут поступать в систему извне (напр.,
за счет притока в проточном реакторе
или путем диффузии
через мембрану)
или образовываться в результате медленных
гомог. р-ций S0iSi
(i=1,2); так же происходит удаление продукта
Р, не влияющего на ход р-ции. S1E,
S1S2E
и S1S1Е
- фермент-субстратные комплексы;
ингибирование
фермента
происходит из-за образования неактивного
комплекса S1S1E.
В этой системе имеется 6 динамич.
переменных: концентрации
субстратов
[S1]
и [S2],
фермента
[Е] и разл. форм фермент-субстратных
комплексов, причем [Е] + [S2E]+[S1S2E]+[S1S1E]=е
- полная концентрация
фермента.
Обычно e<<[S1]
и e<<[S2],
поэтому можно применить квазистационарности
приближение
и представить концентрации
фермент-субстратных комплексов как
алгебраич. ф-ции концентраций
субстратов.
В результате поведение системы можно
описать двумя дифференц. ур-ниями
относительно [S1]
и [S2].
Удобно использовать безразмерные
переменные =[Sl]/K1
и =[S2]/K2
(K1
и К2
-
константы
Михаэлиса), параметры
и 2
-
скорости поступления субстратов,
а также безразмерные комбинации констант
скорости
элементарных стадий
(
и безразмерное время .
Тогда дифференц. ур-ния принимают вид:
Величины
i=i+ii,
наз. характеристич. числами. В неколебат.
устойчивых системах i
отрицательны и действительны (i<0,
i=0).
В этих случаях обычно вместо i
используют времена релаксации
i=1/i.
Если стационарное состояние достаточно
близко к состоянию термодинамич.
равновесия
(выполняются соотношения взаимности
Онсагера, см. Термодинамика
необратимых процессов),
то все i
действительны и отрицательны (теорема
Пригожина).
В этом случае система приближается к
стационарному состоянию без колебаний.
В сильно неравновесных системах i
могут стать комплексными числами, что
соответствует появлению колебаний
около стационарного состояния. При
определенных значениях параметров
сильно неравновесной системы (концентраций
исходных реагентов,
т-ры, давления
и т.д.) стационарное состояние может
потерять устойчивость. Потеря устойчивости
стационарного состояния является
частным случаем бифуркации, т.е. изменения
при определенном (бифуркационном)
значении к.-л. параметра числа или типа
разл. кинетич. режимов системы. Имеется
два простейших случая бифуркации
устойчивого стационарного состояния.
В первом случае одно i
становится положительным. При этом в
точке бифуркации (i=0)
исходно устойчивое состояние становится
неустойчивым или сливается с неустойчивым
стационарным состоянием и исчезает, а
система переходит в новое устойчивое
состояние. В пространстве параметров
в окрестности этой бифуркации существует
область, где система обладает по крайней
мере тремя стационарными состояниями,
из к-рых два устойчивы, а одно неустойчиво.
Во втором случае действит. часть одной
пары
комплексных характеристич. чисел
становится положительной. При этом в
окрестности потерявшего устойчивость
стационарного состояния возникают
устойчивые колебания. После прохождения
точки бифуркации при дальнейшем изменении
параметра количеств, характеристики
колебаний (частота, амплитуда и т.д.)
могут сильно меняться, но качеств. тип
поведения системы сохраняется. В хим.
системах неустойчивости могут возникать
в результате ускорения р-ции ее продуктами
или др. видов автокатализа,
субстратного или перекрестного
ингибирования (см. Ингибиторы),
конкуренции исходных в-в за промежут.
соед. и т.п. В неизотермич. системах
причиной неустойчивости может служить
самоускорение экзотермич. стадий р-ции,
а в электрохим. р-циях экспоненциальная
зависимость скорости р-ции от поляризации
электродов.
Появление простейших неустойчивостей
и соответствующих кинетич. состояний
системы удобно пояснить на примере
ферментативной р-ции с двумя
субстратами
S1
и S2,
один из к-рых, напр. S1,
ингибирует фермент
Е:
S01S1
S02S2
S1+E1
S1E
S1E+S2S1E:P
S1E+S1S1S1E
Субстраты
S1
и S2
могут поступать в систему извне (напр.,
за счет притока в проточном реакторе
или путем диффузии
через мембрану)
или образовываться в результате медленных
гомог. р-ций S0iSi
(i=1,2); так же происходит удаление продукта
Р, не влияющего на ход р-ции. S1E,
S1S2E
и S1S1Е
- фермент-субстратные комплексы;
ингибирование
фермента
происходит из-за образования неактивного
комплекса S1S1E.
В этой системе имеется 6 динамич.
переменных: концентрации
субстратов
[S1]
и [S2],
фермента
[Е] и разл. форм фермент-субстратных
комплексов, причем [Е] + [S2E]+[S1S2E]+[S1S1E]=е
- полная концентрация
фермента.
Обычно e<<[S1]
и e<<[S2],
поэтому можно применить квазистационарности
приближение
и представить концентрации
фермент-субстратных комплексов как
алгебраич. ф-ции концентраций
субстратов.
В результате поведение системы можно
описать двумя дифференц. ур-ниями
относительно [S1]
и [S2].
Удобно использовать безразмерные
переменные =[Sl]/K1
и =[S2]/K2
(K1
и К2
-
константы
Михаэлиса), параметры
и 2
-
скорости поступления субстратов,
а также безразмерные комбинации констант
скорости
элементарных стадий
(
и безразмерное время .
Тогда дифференц. ур-ния принимают вид:
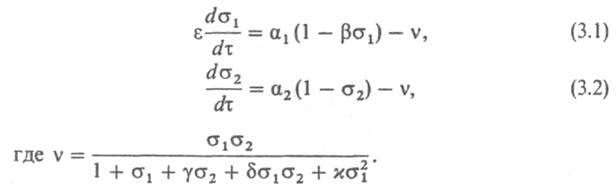 Рассмотрим
случай, когда эта система имеет два
устойчивых стационарных состояния -
бистабильную систему, или триггер. Если
2>>/,
т.е. скорость р-ции S02S2
очень велика по сравнению со скоростью
р-ции S01S1
и скоростью ферментативной р-ции, то
[S2]
постоянна и равна [S02].
В этом случае поведение системы
описывается только одним ур-нием (3.1).
Зависимости dl/d
от
при разных значениях 1
показаны на рис. 1, а. Пунктирные кривые
соответствуют бифуркац. значениям
параметра '1
и :1,
а кривые, заключенные между ними, трижды
пересекают ось абсцисс. Точки пересечения
соответствуют стационарным состояниям
101,
и ,
среднее из к-рых 02
неустойчиво и разделяет области
притяжения устойчивых состояний 101
Рассмотрим
случай, когда эта система имеет два
устойчивых стационарных состояния -
бистабильную систему, или триггер. Если
2>>/,
т.е. скорость р-ции S02S2
очень велика по сравнению со скоростью
р-ции S01S1
и скоростью ферментативной р-ции, то
[S2]
постоянна и равна [S02].
В этом случае поведение системы
описывается только одним ур-нием (3.1).
Зависимости dl/d
от
при разных значениях 1
показаны на рис. 1, а. Пунктирные кривые
соответствуют бифуркац. значениям
параметра '1
и :1,
а кривые, заключенные между ними, трижды
пересекают ось абсцисс. Точки пересечения
соответствуют стационарным состояниям
101,
и ,
среднее из к-рых 02
неустойчиво и разделяет области
притяжения устойчивых состояний 101
 Рис.
1. Ферментативная система с тремя
стационарными состояниями (биохим.
триггер):
зависимость скорости d/d
изменения безразмерной концентрации
субстрата
S1,
от ее значения ()
при разл. скоростях ()
поступления субстрата;
пунктиром обозначены кривые, соответствующие
бифуркац. значениям '1
и ''1;
-
зависимость стационарных значений
от ;
101
и 3
устойчивые, 2
-
неустойчивое стационарные состояния.
Рис.
1. Ферментативная система с тремя
стационарными состояниями (биохим.
триггер):
зависимость скорости d/d
изменения безразмерной концентрации
субстрата
S1,
от ее значения ()
при разл. скоростях ()
поступления субстрата;
пунктиром обозначены кривые, соответствующие
бифуркац. значениям '1
и ''1;
-
зависимость стационарных значений
от ;
101
и 3
устойчивые, 2
-
неустойчивое стационарные состояния.
и
3.
На кривой зависимости стационарной
концентрации
от
(рис. 1, б) область с тремя стационарными
состояниями лежит в интервале ('1,
''1).
При прямом и обратном медленном изменении
параметра
происходит движение системы по различным
траекториям, т.е. гистерезис. Следует
отметить, что описанную бистабильность
можно получить в системе с односубстратной
р-цией, к-рая ведет себя аналогично
двухсубстратной р-ции с фиксир.
концентрацией
одного из субстратов.
Чтобы
система с одной переменной и бистабильностью
стала колебательной, нужно превратить
параметр в медленную переменную. В
ферментативной системе с двумя субстратами
таким параметром, естественно, является
концентрация
второго субстрата
2.
В этом случае для описания системы нужно
использовать оба ур-ния (3). Относительные
изменения концентрации
S2([S2]/[S2])
будут медленными по сравнению с
относительными изменениями Sl,
если [S2]>>[S1].
При переходе к безразмерным параметрам
это условие принимает след, вид: 1~2~1,
<<1.
На фазовой плоскости с координатами
1,
2
поведение системы качественно определяется
взаимным расположением нуль-изоклин-кривых,
на к-рых производные d/d
и dd
равны 0 (рис. 2, а). Точки пересечения
нуль-изоклин соответствуют стационарным
состояниям системы. Пунктиром показано
положение нуль-изоклины d/d=0
при бифуркации, сопровождающейся
возникновением устойчивых колебаний
(автоколебаний) малой амплитуды. Этим
колебаниям соответствует замкнутая
траектория движения системы - т. наз.
предельный цикл. Сплошными линиями
показаны нуль-изоклины в ситуации,
далекой от бифуркации, когда единственное
стационарное состояние системы (точка
О на рис. 2, а) сильно неустойчиво и
окружено предельным циклом ABCD. Движению
системы по этому предельному циклу
соответствуют автоколебания концентраций
и 2
с большой амплитудой (см. рис. 2, б).
 Рис.
2. Автоколебания (устойчивые колебания)
в модельной ферментативной системе:
a-фазовая плоскость в координатах -2
с нуль-изоклинами d/d=0,
d2/d=0;
пунктиром показано положение нуль-изоклины
d/d=0,
соответствующее колебат. бифуркации,
и малый предельный цикл, окружающий
потерявшее устойчивость стационарное
состояние О, ABCD большой предельный цикл;
б - автоколебания концентраций
и 2,
соответствующие большому предельному
циклу ABCD.
Рис.
2. Автоколебания (устойчивые колебания)
в модельной ферментативной системе:
a-фазовая плоскость в координатах -2
с нуль-изоклинами d/d=0,
d2/d=0;
пунктиром показано положение нуль-изоклины
d/d=0,
соответствующее колебат. бифуркации,
и малый предельный цикл, окружающий
потерявшее устойчивость стационарное
состояние О, ABCD большой предельный цикл;
б - автоколебания концентраций
и 2,
соответствующие большому предельному
циклу ABCD.
В
ходе колебательных реакций
наблюдались периодич. колебания разл.
формы: синусоидальные, пилообразные,
прямоугольные и т.д.; модулированные,
квазипериодические и стохастические.
Периоды большинства колебательных
реакций
лежат в диапазоне от долей секунды до
десятков минут. К жидкофазным колебательным
реакциям
относятся, напр., диспропорционирование
Н2О2
и S2O42-,
окисление
разл. в-в галогенкислородными соед.,
окисление
углеводородов
и сульфидов
кислородом.
Хорошо изучена Белоусова - Жаботинского
реакция,
идущая в водном р-ре, где НВrO3
при катализе
ионами
металлов
переменной валентности
окисляет разл. орг. соед., в частности
малоновую к-ту.
Газофазные
колебательные реакции
обнаружены и исследованы при окислении
паров
фосфора,
углеводородов,
СО и др. соединений. Во всех случаях
существенны как объемные стадии р-ции,
так и обрыв и зарождение цепей на стенках
реактора, а также ускорение р-ций за
счет разогрева системы в результате
экзотермич.
стадий (тепловой автокатализ).
Возможны чисто термокинетич. автоколебания,
когда тепловой автокатализ
является единств, причиной неустойчивости.
Простейшая модель термокинетич. колебаний
в проточном реакторе имеет вид: В0:В:Р+Q.
Здесь в-во В поступает в проточный
реактор идеального смешения,
где происходит мономолекулярная
экзотермич. р-ция распада; выделяющееся
тепло отводится через стенку реактора.
Кинетика этой р-ции описывается двумя
дифференц. ур-ниями относительно
концентрации
В и т-ры Т внутри реактора:
 где
[В0]
- приведенная концентрация
на входе в реактор, Т0
-
т-ра стенки реактора, k - коэф. скорости
обновления реакц. смеси в реакторе, h -
коэф. скорости теплообмена,
Q - тепловой эффект р-ции, Ср
- теплоемкость
при постоянном давлении,
-
плотность, Е и А - энергии
активации
и предэкспоненциальный множитель р-ции
соотв., R - газовая
постоянная.
В этой системе саморазогрев ускоряет
р-цию, что приводит к исчерпанию В в
реакторе и замедлению р-ции; затем
концентрация
В растет вследствие его поступления в
реактор и цикл повторяется.
Гетерог.
колебательные реакции
имеют место при окислении
СО, Н2,
NH3,
С2Н4,
СН3ОН
на катализаторах
платиновой группы. Часто колебания
наблюдаются при растворении
или осаждении
металлов
на границе металл
- раствор.
Обычно эти колебательные реакции
связаны с электрохим. р-циями образования
новой фазы. На рис. 3, а-е показаны примеры
различных
где
[В0]
- приведенная концентрация
на входе в реактор, Т0
-
т-ра стенки реактора, k - коэф. скорости
обновления реакц. смеси в реакторе, h -
коэф. скорости теплообмена,
Q - тепловой эффект р-ции, Ср
- теплоемкость
при постоянном давлении,
-
плотность, Е и А - энергии
активации
и предэкспоненциальный множитель р-ции
соотв., R - газовая
постоянная.
В этой системе саморазогрев ускоряет
р-цию, что приводит к исчерпанию В в
реакторе и замедлению р-ции; затем
концентрация
В растет вследствие его поступления в
реактор и цикл повторяется.
Гетерог.
колебательные реакции
имеют место при окислении
СО, Н2,
NH3,
С2Н4,
СН3ОН
на катализаторах
платиновой группы. Часто колебания
наблюдаются при растворении
или осаждении
металлов
на границе металл
- раствор.
Обычно эти колебательные реакции
связаны с электрохим. р-циями образования
новой фазы. На рис. 3, а-е показаны примеры
различных
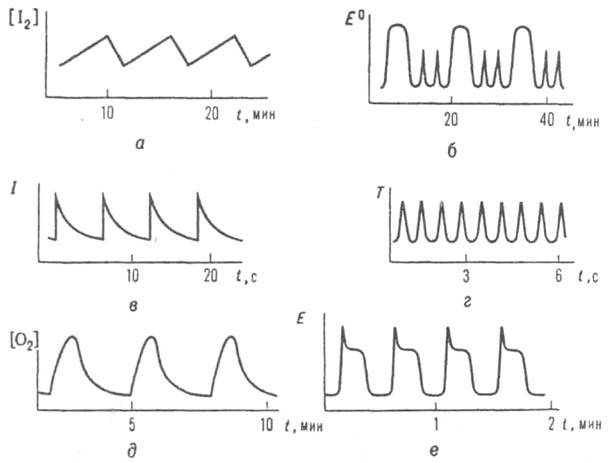 Рис.
3. Автоколебания в разл. хим. системах:
а колебания концентрации
I2
при разложении Н2О2
в присут. IО3;
6 колебания окислит.-восстановит.
потенциала E0
р-ра при окислении
S2O32-
хлоритом;
в колебания интенсивности I хемилюминесценции
при газофазном окислении
СО; г - колебания т-ры T0
при газофазном окислении
СН3СНО;
д - колебания конц. О2
при окислении
Н2
на Ni; е - колебания электродного
потенциала
E при растворении
Fe в HNO3.
Рис.
3. Автоколебания в разл. хим. системах:
а колебания концентрации
I2
при разложении Н2О2
в присут. IО3;
6 колебания окислит.-восстановит.
потенциала E0
р-ра при окислении
S2O32-
хлоритом;
в колебания интенсивности I хемилюминесценции
при газофазном окислении
СО; г - колебания т-ры T0
при газофазном окислении
СН3СНО;
д - колебания конц. О2
при окислении
Н2
на Ni; е - колебания электродного
потенциала
E при растворении
Fe в HNO3.
автоколебат. хим. систем. Колебательная реакция может протекать и в распределенной системе, где имеется диффузионная связь между отдельными элементами пространства, напр. при р-циях в тонком слое неперемешиваемой жидкости. В таких случаях возникают бегущие концентрац. волны. Колебания могут возникать при работе проточных реакторов (напр., при полимеризации этилена, окислении СО). Обычно они вредны, снижают однородность продукта, приводят к аварийным ситуациям. Однако в ряде случаев проведение р-ции в колебат. режиме может быть полезным. Напр., средняя скорость каталитич. окисления SO2 на V2O5 возрастает в колебат. режиме на 15%; в ряде процессов полимеризации в результате колебаний скорости подачи мономера снижается полидиспeрсность продукта. Колебательные реакции лежат в основе ряда важнейших биол. процессов: генерации биоритмов, мышечного сокращения и т.д. Важнейшая биол. колебательная реакция- генерация нервных импульсов, вызываемая изменением проницаемости трансмембранных ионпроводящих каналов.
Влияние температуры на скорость химической реакции
Влияние температуры на скорость
Правило
Вант-Гоффа: при повышении Т на
![]() скорость
хим. реакции увеличивается в 2-4 раза.
Математически это правило можно записать:
скорость
хим. реакции увеличивается в 2-4 раза.
Математически это правило можно записать:
![]() ,
,
![]() ,
,
![]()
![]() -
температурный коэффициент хим. реакции.
Правило Вант-Гоффа является приближённым
и его обычно используют для приблизительно
оценки скорости при изменении температуры.
Более точным является уравнение
Аррениуса, по которому:
-
температурный коэффициент хим. реакции.
Правило Вант-Гоффа является приближённым
и его обычно используют для приблизительно
оценки скорости при изменении температуры.
Более точным является уравнение
Аррениуса, по которому:![]() .
Они могут быть вычислены по значению
констант скорости при 2-х различных Т.
При
.
Они могут быть вычислены по значению
констант скорости при 2-х различных Т.
При
![]() :
:
![]() (1).
При
(1).
При
![]() :
:
![]() (2).
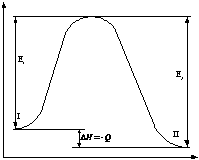
Вычитая из (1) (2) получаем
(2).
Вычитая из (1) (2) получаем
![]() .
Отсюда можно выразить А. Зная А, по
уравнению (1) или (2) вычисляют В. Уравнение
Аррениуса может быть получено т/д-им
выводом из уравнения изобары (изохоры)
хим. реакции. Опуская индексы,
характеризующие условия протекания
реакции, это уравнение записывается:
.
Отсюда можно выразить А. Зная А, по
уравнению (1) или (2) вычисляют В. Уравнение
Аррениуса может быть получено т/д-им
выводом из уравнения изобары (изохоры)
хим. реакции. Опуская индексы,
характеризующие условия протекания
реакции, это уравнение записывается:
![]() ,
,
![]() ,
где
,
где
![]() и
и
![]() -
константы скорости прямой и обратной
реакции. Учитывая эти уравнения можно
записать:
-
константы скорости прямой и обратной
реакции. Учитывая эти уравнения можно
записать:
![]() .
Представим тепловой эффект реакции Q
как разность 2-х энергетических величин:
.
Представим тепловой эффект реакции Q
как разность 2-х энергетических величин:
![]() .
Тогда последнее уравнение можно записать
в виде:
.
Тогда последнее уравнение можно записать
в виде:
![]() .
С точностью до некоторой постоянной
величины можно записать:
.
С точностью до некоторой постоянной
величины можно записать:
![]() ,
,
![]() .
Опыт показывает что
.
Опыт показывает что
![]() .
Отбрасывая индексы, последнее уравнение
записывается:
.
Отбрасывая индексы, последнее уравнение
записывается:
![]() (1),
где К – константа скорости хим. реакции.
Энергетическая величина Е в этом
уравнение называется энергией активации.
Полученное уравнение описывает
зависимость К хим. реакции от температуры.
Разделив переменные и проинтегрировав,
получим:
(1),
где К – константа скорости хим. реакции.
Энергетическая величина Е в этом
уравнение называется энергией активации.
Полученное уравнение описывает
зависимость К хим. реакции от температуры.
Разделив переменные и проинтегрировав,
получим:
![]() ,
,
![]() (2).
(2).
Уравнение
(2) по форме походит на уравнение Аррениуса,
интегрируя (2) получим:
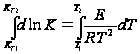 ,
,
![]() (3).
(3).
Уравнение
используют либо для вычисления энергии
активации по известным константам
скорости при двух температурах, либо
для вычисления константы скорости
реакции при неизменной температуре,
если известна энергия активации. Для
большинства хим. реакций энергия
активации определяется в пределах
![]() .
Физический смысл энергии активации
раскрывается в теории химической
кинетики, её можно определить как
некоторый избыток энергии по сравнению
со средним значением для денных условий,
которыми должны обладать молекулы чтобы
вступить в хим. реакцию. Уравнение (2)
чаще представляют в виде:
.
Физический смысл энергии активации
раскрывается в теории химической
кинетики, её можно определить как
некоторый избыток энергии по сравнению
со средним значением для денных условий,
которыми должны обладать молекулы чтобы
вступить в хим. реакцию. Уравнение (2)
чаще представляют в виде:
![]() .
При этом
.
При этом
![]() называют
предэкспоненциальным множителем. Связь
энергии активации с тепловым эффектом
можно проиллюстрировать с помощью
представлению о энергетическом барьере,
который разделяет начальное и конечное
состояние системы. I и II – уровни энергии
вещ-в исходных и продуктов реакции.
называют
предэкспоненциальным множителем. Связь
энергии активации с тепловым эффектом
можно проиллюстрировать с помощью
представлению о энергетическом барьере,
который разделяет начальное и конечное
состояние системы. I и II – уровни энергии
вещ-в исходных и продуктов реакции.
![]() -
энергия активации прямой реакции.
-
энергия активации прямой реакции.
![]() -
энергия активации обратной реакции.
Избыток энергии реагирующих молекул,
названный энергией активации, необходим
для преодоления отталкивания электронных
облаков взаимодействующих молекул при
их столкновении, и для разрыва старых
связей молекул. Уравнение Аррениуса
справедливо в области невысоких
температур; при достаточно высоких
температурах константа скорости
перестаёт зависеть от температуры.
-
энергия активации обратной реакции.
Избыток энергии реагирующих молекул,
названный энергией активации, необходим
для преодоления отталкивания электронных
облаков взаимодействующих молекул при
их столкновении, и для разрыва старых
связей молекул. Уравнение Аррениуса
справедливо в области невысоких
температур; при достаточно высоких
температурах константа скорости
перестаёт зависеть от температуры.
Правило Вант-Гоффа — эмпирическое правило, позволяющее в первом приближении оценить влияние температуры на скорость химической реакции в небольшом температурном интервале (обычно от 0 °C до 100 °C). Я. Х. Вант-Гофф на основании множества экспериментов сформулировал следующее правило:
-
При повышении температуры на каждые 10 градусов константа скорости гомогенной элементарной реакции увеличивается в два — четыре раза.
Уравнение, которое описывает это правило следующее:
![]()
где
![]() —
скорость реакции при температуре
—
скорость реакции при температуре
![]() ,
,
![]() —
скорость реакции при температуре
—
скорость реакции при температуре
![]() ,
,
![]() —
температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
—
температурный коэффициент реакции
(если он равен 2, например, то скорость
реакции будет увеличиваться в 2 раза
при повышении температуры на 10 градусов).
Следует помнить, что правило Вант-Гоффа применимо только для реакций с энергией активации 60-120 кДж/моль в температурном диапазоне 10-400oC. Правилу Вант-Гоффа также не подчиняются реакции, в которых принимают участие громоздкие молекулы, например белки в биологических системах. Температурную зависимость скорости реакции более корректно описывает уравнение Аррениуса.
Из уравнения Вант-Гоффа температурный коэффициент вычисляется по формуле:
![]()
Порядок химической реакции
Порядок реакции по данному веществу — показатель степени при концентрации этого вещества в кинетическом уравнении реакции.
[Править] Реакция нулевого порядка
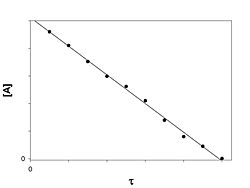
![]()
График зависимости концентрации реагента A в реакции A → B от времени для нулевого порядка реакции
Кинетическое уравнение имеет следующий вид:
V0 = k0
Скорость реакции нулевого порядка постоянна во времени и не зависит от концентраций реагирующих веществ. Нулевой порядок характерен, например, для гетерогенных реакций в том случае, если скорость диффузии реагентов к поверхности раздела фаз меньше скорости их химического превращения.
[Править] Реакция первого порядка

График зависимости концентрации реагента A для первого порядка реакции
Кинетическое уравнение реакции первого порядка:
![]()
Приведение уравнения к линейному виду даёт уравнение:
![]()
Константа скорости реакции вычисляется как тангенс угла наклона прямой к оси времени:
k1 = − tgα
Период полупревращения:
![]()
[Править] Реакция второго порядка
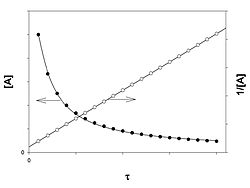
График зависимости концентрации реагента A для второго порядка реакции
Для реакций второго порядка кинетическое уравнение имеет следующий вид:
![]()
или
![]()
В первом случае скорость реакции определяется уравнением
![]()
Линейная форма уравнения:
![]()
Константа скорости реакции равна тангенсу угла наклона прямой к оси времени:
k2 = − tgα
![]()
Во втором случае выражение для константы скорости реакции будет выглядеть так:
![]()
Период полупревращения (для случая равных начальных концентраций!):
![]()
Энергия активации в химии и биологии — минимальное количество энергии, которое требуется сообщить системе (в химии выражается в джоулях на моль), чтобы произошла реакция. Термин введён Сванте Августом Аррениусом в 1889. Типичное обозначение энергии реакции Ea.
Энергия активации в физике — минимальное количество энергии, которое должны получить электроны донорной примеси, для того чтобы попасть в зону проводимости.
В химической модели, известной как Теория активных соударений (ТАС), есть три условия, необходимых для того, чтобы произошла реакция:
Молекулы должны столкнуться. Это важное условие, однако его не достаточно, так как при столкновении не обязательно произойдёт реакция.
Молекулы должны обладать необходимой энергией (энергией активации). В процессе химической реакции взаимодействующие молекулы должны пройти через промежуточное состояние, которое может обладать большей энергией. То есть молекулы должны преодолеть энергетический барьер; если этого не произойдёт, реакция не начнётся.
Молекулы должны быть правильно ориентированы относительно друг друга.
При низкой (для определённой реакции) температуре большинство молекул обладают энергией меньшей, чем энергия активации, и неспособны преодолеть энергетический барьер. Однако в веществе всегда найдутся отдельные молекулы, энергия которых значительно выше средней. Даже при низких температурах большинство реакций продолжают идти. Увеличение температуры позволяет увеличить долю молекул, обладающих достаточной энергией, чтобы преодолеть энергетический барьер. Таким образом повышается скорость реакции.
|
|