

Фарфоровий посуд:
Фарфоровий посуд порівняно зі скляним більш міцний та термостійкий (мал.1.13). Можна випарювати розчини досуха та спалювати речовини (при t ≤ 1200 С). Проте у фарфоровому посуді не можна проводити сплавлення з лугами та працювати з плавиковою кислотою.
Фарфорові чашки (для випарювання розчинів і просушування твердих порошкоподібних речовин);
Фарфорові тиглі (для прожарювання, спалювання органічних і неорганічних речовин до температури 1000-11000С). Тиглі для прожарювання випускають різних розмірів за висотою та діаметром.
Ступки: фарфорові, скляні, агатові.
Використовують для подрібнення і розтирання твердих речовин.
Вимірювальний посуд (товстостінний скляний посуд):
Мірні циліндри ‑ скляні товстостінні посудини з поділками на зовнішній стінці, вказують об’єм в мілілітрах. Використовують для вимірювання об’ємів рідин з точність 0,2 ‑ 0,3 ціни поділки (мал.1.14).
Піпетки (піпетки Мора) – скляні трубки певного об’єму від 1 до 100мл (мал.1.15). На градуйованих піпетках нанесені поділки з ціною 0,1мл. Використовують для точного вимірювання об’єму рідини. Для заповнення піпетки використовують «гумову грушу».
Мірні колби – скляні плоскодонні колби різної ємності з притертими скляними або підібраними гумовими або поліетиленовими корками (мал.1.16). На горлі колби є риска, до рівня якої необхідно довести об’єм розчину, щоб одержати об’єм розчину, вказаний на колбі в мілілітрах. Використовують для виготовлення розчинів заданої точної концентрації.
Бюретки використовують для точного відліку об’єму реактиву, для титрування в об’ємному аналізі. Об’ємні бюретки – скляні трубки з краном або витягнутим нижнім кінцем. На зовнішній стінці бюретки по всій довжині нанесені поділки з ціною 0,1мл (мал.1.17).
Бюретку закріплюють у лабораторному штативі в лапках або спеціальних утримувачах (мал.1.18). Після роботи бюретку промивають водою та вміщують в штатив, перевернувши відкритим кінцем вгору. У бюреток з краном необхідно витягнути кран, обернути фільтрувальним папером і знову вставити в бюретку. Бюретки заповнюють рідиною через лійку, потім заповнюють частину бюретки нижче крану або зажиму для видалення бульбашок повітря. Рівень рідини встановлюється за нижнім меніском.
Використовують бюретки з автоматичним встановленням рівня (розчин піднімається вгору до верхньої риски, а надлишок його зливається в посудину через спеціальну трубку). Для одержання більш точних результатів у бюретку вміщують скляний поплавок, що дозволяє підвищити точність відліку рівня рідини. Для проведення аналізів напівмікрометодом використовують мікробюретки з ціною поділки 0,01мл.
Металічне обладнання
Універсальні штативи, треноги, зажими, тигельні щипці, пінцети, тиглі металічні, платиновий посуд, утримувачі для пробірок, фарфорових чашок та стаканів.
Металеве обладнання слід берегти від корозії та механічних пошкоджень.
|
Мал.1.1. Пробірки: а – лабораторна звичайна; б – із загнутими краями; в – мірна зі шліфом; г – конічна з поділками |
|
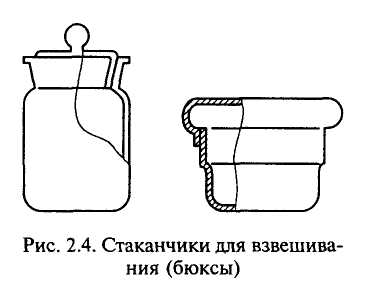
Мал.1.2. Хімічний стакан |
|
|
Мал.1.3. Конічна колба Ерленмейера (а) та Бунзена (б) для вакуумного фільтрування |
|
Мал.1.4. Лійки: а – конічна звичайна; б – з подовженим кінцем; в – для твердих речовин |
|
|
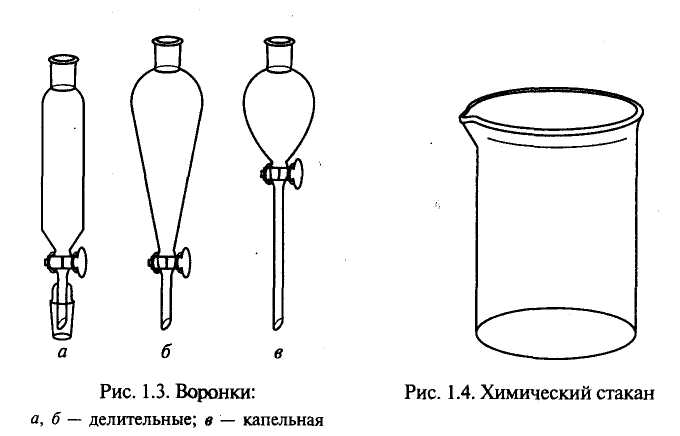
Мал.1.5. Лійки: а, б – ділильні; в ‑ крапельні |
|
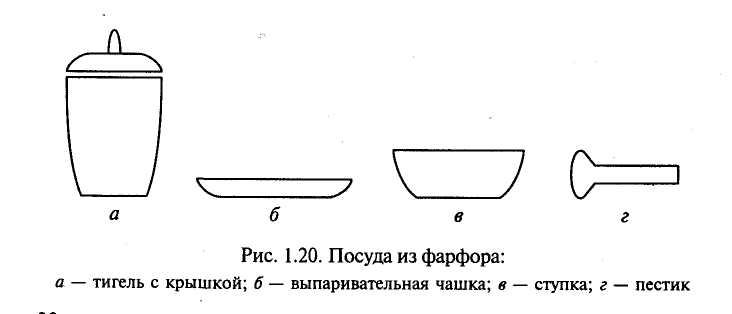
Мал.1.6. Стаканчики для зважування (бюкси) |
|
|
Мал.1.7. Лійка Бюхнера |
|
Мал.1.8. Колби круглодонні |
|
|
Мал.1.9. Одно-, двох- та трьохгорлі колби для дистиляції та синтезу речовин |
|
Мал.1.10. Колба В’юрца для перегонки |
|
|
Мал.1.11. Звичайний ексикатор (а) та вакуум-ексикатор (б) |
|
Мал.1.12. Холодильники: а – прямий (Лібіха); б – кульковий; в – д ‑ змійовикові |
|
|
Мал. 1.13.Посуд з фарфору: а – тигель з кришкою; б – випарювальна чашка; в – ступка; г – пестик. |
|
Мал.1.14. Мірний циліндр |
|
|
Мал.1.15. Мірні піпетки (піпетки Мора) |
|
Мал.1.16. Мірна колба |
|
|
Мал.1.17. Бюретка з краном |
|
Мал.1.18. Штатив лабораторний |
|









Прилади та оснащення сучасної хімічної лабораторії:
Нагрівальні (спиртівка, плитка);
Сушильна шафа (для висушування речовин при температурі (100-1100С);
Муфельна піч;
Ваговимірювальне обладнання (технохімічні та аналітичні терези).
В лабораторіях для зважування речовин користуються технохімічними терезами, на яких можна зважувати з точністю до 0,01г, і аналітичними терезами (точність зважування 0,0001г).
Прилади для струшування та перемішування (магнітні мішалки);
Центрифуга;
Прилади для проведення фільтрування під високим тиском або розрідженням;
Прилади для відбору проб досліджуваного матеріалу;
Дистилятор;
Гомогенізатор (для подрібнення харчового матеріалу взятого для дослідження);
Рефрактометр (для визначення сухих речовин);
Фотоелектроколориметр (фотометр, спектрофотометр);
Іономір (рН-метр);
Поляриметр;
Аналізатор вологості (апарат Чижової);
Прилад для люмінесцентного аналізу;
Турбодиметр (для визначення ступеня каламутності розчинів);
Лабораторні вимірювальні системи для проведення різноманітних визначень електрохімічними методами;
Титратори (для проведення кількісного аналізу об’ємними методами);
Портативні лабораторії, оснащені експрес-тестами, ампулами з розфасованими реактивами, портативними приладами для проведення досліджень в польових умовах.
Хроматограф.
Лекція №2. Закон дії мас як теоретична основа аналітичної хімії
Основні питання:
Закон дії мас.
Хімічна рівновага. Константа рівноваги.
Способи заміщення рівноваги.
Застосування закону дії мас до різних типів хімічних реакцій.
Закон діючих мас (Гульдберг і Вааге, 1867) – основний закон хімічної кінетики: швидкість хімічної реакції пропорційна добуткові концентрацій реагентів.
У загальному випадку для реакції: а А + b В ↔ с С + d D
υ = κ ∙САа ∙ СВb , де
κ – константа швидкості реакції (швидкість реакції при концентраціях реагентів 1моль/л), характеризує здатність реагентів до взаємодії.
СА і СВ – концентрації реагентів;
а, b– коефіцієнти в рівнянні реакції.
Константа швидкості реакції залежить від:
• природи реагентів;
• температури;
• наявності каталізаторів.
Константа швидкості реакції не залежить від:
• концентрацій речовин;
• часу.

Стан реагуючої системи, при якому швидкості прямої та зворотної реакцій стають однаковими, називається хімічною рівновагою. Хімічна рівновага має динамічний характер (кількості вихідних речовин і продуктів не змінюються).
Концентрації всіх реагуючих речовин в стані рівноваги називаються рівноважними концентраціями, [ ].
Кількісною характеристикою стану хімічної рівноваги є константа рівноваги (Крівн).
Константа рівноваги – це відношення добутку концентрацій продуктів реакції до добутку концентрацій вихідних речовин після досягнення рівноваги.
Математичний вираз закону діючих мас для стану рівноваги (можна застосовувати для рівноважного стану будь-якої оборотної реакції незалежно від механізму її перебігу).
Хімічна реакція в загальному вигляді: а А + в В ↔ с С + d D
Крівн.
=
![]() =
=
![]() ,
де
,
де
[А] і [В] – рівноважні концентрації вихідних речовин;
[С] і [Д] – рівноважні концентрації продуктів;
а, в, с, d – стехіометричні коефіцієнти в рівнянні реакції;
κ1 – константа швидкості прямої реакції;
κ2 – константа швидкості зворотної реакції.
К рівноваги свідчить:
відношення добутків рівноважних концентрацій продуктів і вихідних речовин у степенях, що дорівнюють їх стехіометричним коефіцієнтам, є величиною сталою;
про глибину проходження реакції (у скільки разів пряма реакції відбувається швидше, ніж зворотна, за однакової температури й при концентраціях, що дорівнюють одиниці).
Для
необоротних реакцій Крівн
→
![]() ;
К→ 0 (відсутність хімічної взаємодії).
;
К→ 0 (відсутність хімічної взаємодії).
Змінюючи умови, можна перевести систему з одного рівноважного стану в інший.
Перехід системи від одного стану рівноваги до іншого – зміщення хімічної рівноваги.
Принцип Ле Шательє (принцип рухомої рівноваги) (1884): якщо на систему, що перебуває в стані рівноваги, впливати ззовні, то рівновага зміщується у бік тієї реакції, що послаблює цей вплив.
Вплив концентрації на стан рівноваги
Реакція |
Концентрація реагентів |
Концентрація продуктів |
Пряма |
збільшення |
зменшення |
Зворотна |
зменшення |
збільшення |
Вплив температури на стан рівноваги
Пряма реакція |
Підвищення температури |
Зниження температури |
Екзотермічна |
рівновага зміщується вліво (відбувається зворотна реакція) |
рівновага зміщується вправо (відбувається пряма реакція) |
Ендотермічна |
рівновага зміщується вправо (відбувається пряма реакція) |
рівновага зміщується вліво (відбувається зворотна реакція) |
Вплив тиску на стан рівноваги
Пряма реакція |
Збільшення тиску |
Зменшення тиску |
Зі збільшенням об’єму |
рівновага зміщується вліво (відбувається зворотна реакція) |
рівновага зміщується вправо (відбувається пряма реакція) |
Зі зменшенням об’єму |
рівновага зміщується вправо (відбувається пряма реакція) |
рівновага зміщується вліво (відбувається зворотна реакція) |
Застосування принципу Ле Шательє

Застосування закону дії мас до різних типів хімічних реакцій
№п/п |
Тип реакції |
Рівняння реакції |
Математичний вираз |
Назва константи рівноваги |
1. |
Дисоціація слабкого електроліту |
АаВb ↔ аАb+ bВа- |
Кдис.
=
|
константа дисоціації, Кдис |
2. |
Дисоціація малорозчинного електроліту |
АаВb ↔ аАb+ + bВа- |
ДР(АаВb)=[Аb+]а·[Ва-]b |
добуток розчинності, ДР (АаВb) |
3. |
Дисоціація води |
Н2О↔Н++ОН- |
Кω=[Н+]·[ОН-]=10-14 |
іонний добуток води, Кω |
4. |
Гідроліз солей |
А- + H+OH- ↔ HCN + OH- К+ + H+OH- ↔ КOH + H+ К+ + А- ↔ КОН + НА |
Кг
=
Кг
=
Кг
=
|
константа гідролізу, Кг |
Лекція №3. Розчини. Кількісний склад розчинів
Основні питання:
Розчини. Класифікація розчинів.
Способи вираження кількісного складу розчинів.
Найбільш поширеним фізико-хімічними системами, з якими зустрічаються в повсякденному житті, є розчини.
Розчини – це однорідні (гомогенні) системи, що складаються з двох і більше компонентів (складових частин) і продуктів їх взаємодії.
Розчинник – компонент розчину, що кількісно переважає і зберігає свій агрегатний стан при утворенні розчину.
Розчинена речовина – компонент розчину, що міститься в меншій кількості.


Кількісний склад розчину визначається відносним вмістом кожного з його компонентів. Фізична величина, яка визначає кількісний склад розчину, називається концентрацією.
ІUРАС рекомендує використовувати кілька способів вираження кількісного складу розчинів, які ґрунтуються на сталості маси розчину, розчиненої речовини, розчинника чи об’єму розчину.





Лекція №4. Рівноваги в гомогенних системах
Основні питання:
Розчини електролітів: теорія електролітичної дисоціації, теорія сильних електролітів.
Кількісні характеристики процесу дисоціації: ступінь дисоціація, константа дисоціації.
Йонний добуток води. Водневий показник. Класифікація розчинів за значенням рН.
Електроліти – речовини, розплави або розчини яких, проводять електричний струм.
Ступінь дисоціації (α) – відношення числа частинок електроліту, що розпалися на іони, до загального числа його частинок у розчині.
α
=
![]()
α = 1 – дисоціація повна; α = 0 – дисоціація відсутня; 0< α < 1 – часткова дисоціація.
Ступінь дисоціації залежить від:
природи електроліту та розчинника;
концентрації електроліту (С↓, α ↑);
температури (Т↑, α ↑).
Експериментальне визначення ступеня дисоціації (α) проводиться:
за значенням ізотонічного коефіцієнта;
вимірюванням електропровідності розчинів різних концентрацій.
|
Залежно від значення ступеня дисоціації електроліти поділяють на: |
|
||
|---|---|---|---|---|
|
Сильні електроліти (α>0,3) – повністю дисоційовані на іони у розчинах будь – яких концентрацій. Належать: розчинні солі, гідроксиди лужних і лужноземельних металів, мінеральні кислоти (HCl, HNO3, H2SO4, HСlO4) |
Слабкі електроліти (α < 0,03) – частково дисоціюють на іони у розчинах будь-яких концентрацій. Належать: основи, солі (погано розчинні), кислоти (H2S, H3BO3, H2CO3, HCN, H2SO3, H2SiO3, HNO2). |
|
|
Сильні електроліти |
Слабкі електроліти |
|||
Теорія сильних електролітів (Дебай, Гюккель, 1923): особливості розчинів сильних електролітів зумовлені наявністю в розчинах міжіонних взаємодій (посилюються зі збільшенням концентрації електроліту).
Результат – зменшення електропровідності розчину (α < 1). Поняття активності іонів (Льюїс, 1907) введено для характеристики властивостей і стану іонів у розчинах сильних електролітів. Активність (а) – це реальна концентрація з урахуванням взаємодії іонів: а = f · с , де f – коефіцієнт активності, характеризує ступінь відхилення властивостей, визначається експериментально (f < 1)
f залежить від:
|
Дисоціація слабкого електроліту: КА ↔ К+ + А- Кдис.
= Кдис. – константа дисоціації, характеризує здатність електроліту до дисоціації (чим більше її значення, тим сильніший електроліт). При ступінчастій дисоціації:
К2А
↔ К+
+ КА-;
К1=
КА-
↔ К+
+ А2-;
К2
=
Сумарна реакція:
К2А
↔ 2К+
+ А2-;
К =
К – загальна константа дисоціації; К1 – ступінчаста константа дисоціації (І стадія); К2 – ступінчаста константа дисоціації (ІІ стадія), причому К1> К2 , К = К1·К2 Кдис. не залежить: від концентрації електроліту в розчині. За величиною Кдис. слабкі електроліти поділяють:
Увага! Основні положення теорії електролітичної дисоціації стосуються електролітів, які зазнають неповної дисоціації. Оборотний характер процесу дисоціації, його ступінчастість втрачають зміст при наявності в розчині сильного електроліту (α =1). Основні положення ТЕД (Арреніус, 1887)
|
|||
 біохімічних дослідженнях, змінюючи
іонну силу електролітів, можна
збільшити або зменшити розчинність
білків і амінокислот, що використовуються
для їх розділення.
біохімічних дослідженнях, змінюючи
іонну силу електролітів, можна
збільшити або зменшити розчинність
білків і амінокислот, що використовуються
для їх розділення.
Розрізняють:
Загальну кислотність (лужність) ‑ число моль еквівалентів кислоти (лугу), що містяться в 1л розчину (відповідає загальній концентрації кислоти або лугу).
Активну кислотність (лужність) – концентрація вільних гідратованих іонів Н+ (або ОН -).
Сильні електроліти рН= -lg[Н+] – для сильної кислоти; рН=14 ‑ рОН – для сильної основи; рОН= ‑ lg[ОН-] – гідроксидний показник; Загальна кислотність (лужність) дорівнює активній кислотності (лужності). Загальна кислотність визначається титруванням. Активна кислотність визначається вимірюванням рН. |
|
Слабкі електроліти рН= ‑ ½ рКa – ½ lgСа – для слабкої кислоти, де Ка і Са – константа іонізації та концентрація слабкої кислоти. рН= ‑ ½ рКb– ½ lgСb – для слабкої основи, де Кв і Св – константа іонізації та концентрація слабкої основи. рКа = ‑ lgKa , де Ка – константа іонізації кислоти; рКb= ‑ lgKb, де Кb– константа іонізації основи. У розчині слабкої кислоти: [Н+]<Са У розчині слабкої основи: [ОН-]<Сb Загальна (лужність) відрізняється від активної кислотності (лужності). |
До складу шлункового соку входять кислоти різної сили, але його кислотність визначається тільки концентрацією іонів водню. Тому безпосереднє вимірювання рН і титрування шлункового соку дадуть різні результати.
Кислотність молока виражають в одиницях титрованої кислотності (у градусах Тернера) і величиною рН при 20оС.
Титрована кислотність свіжовидоєного молока становить 16-18оТ, зумовлена кислими солями – дигідрофосфатами і і дигідроцитратами, казеїном і білками сироватки, вуглекислим газом, кислотами (молочною, лимонною, аскорбіновою, вільними жирними), є критерієм оцінки якості молока при його заготівлі.
При зберіганні сирого молока титрована кислотність зростає відповідно із розвитком у ньому молочнокислих бактерій, які перетворюють лактозу в молочну кислоту. Підвищення кислотності – небажаний процес, знижується стійкість білків молока до нагрівання. Молоко з кислотністю 21оТ приймають як несортове.
Активна кислотність молока – рН ‑ зумовлена дисоціацією кислот і кислих солей, становить 6,5 (відносно стійка завдяки буферній системі). При розвитку в молоці молочнокислих бактерій суттєво зростає титрована кислотність, рН практично не змінюється.
Електрохімічне
визначення рН
(за допомогою рН-метра) – найбільш
точний метод. Базується на вимірюванні
електрорушійної сили (ЕРС) досліджуваного
розчину, що виникає між скляним електродом
(чутливий до концентрації Н+)
та електродом порівняння. Скляний
електрод:
тонкостінна скляна колба з впаяною в
неї срібною дротиною, що заповнена
насиченим розчином NaCl
в 0,1Н НСl
(індикаторний). Хлорсрібний
– електрод порівняння (зовнішній) Схема:
(-)Ag/AgCl,
HCl
/ мембрана / розчин з рНх
/КСl,
AgCl
/ Ag(+)
Переваги
скляних електродів: широкий
діапазон рН в різних середовищах; не
чутливі до окислювально-відновних
процесів; індиферентні
до ПАР і білків.
ЕРС
вимірюється вольтметром, якщо шкала
потенціометра проградуйована в одиницях
рН – прилад рН-метр (рН-340, рН-262, рН-673,
П-261, іономір ЕВ-74), то результат
вимірювання - рН досліджуваного розчину
(не потребує побудови калібрувального
графіка). калібрування
скляного електрода за серією буферних
розчинів; побудова
калібрувального графіка у координатах
Е – рН; вимірявши
Ех
кола з досліджуваним розчином, за
калібрувальним графіком знаходять
рНх
цього розчину. Індикатори
– речовини, що здатні змінювати
забарвлення залежно від ступеня активної
кислотності (лужності) середовища. Універсальні
індикатори
– суміш звичайних індикаторів, мають
вигляд: індикаторного паперу або
розчину.
Застосування:
для швидкого та наближеного визначення
рН розчинів.
Базується
на порівнянні забарвлення досліджуваної
рідини з індикатором із забарвленням
цього ж індикатора в стандартному
розчині. Прилад
Міхаеліса:
набір індикаторів, комплект стандартних
розчинів, компаратор, 4 стандартні
пробірки. Застосування:
для визначення рН слабкозабарвлених
і каламутних рідин.
Визначення рНх за допомогою калібрувального графіка:
Індикаторний метод
Спосіб Міхаеліса

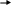
Лекція №5. Буферні розчини. Рівноваги у розчинах солей, що гідролізують
Основні питання:
1. Буферні розчини. Роль буферних систем.
Рівноваги у розчинах солей, що гідролізують. Кількісні характеристики гідролізу.
Буферні розчини
(здатні зберігати значення рН при розведенні або при додаванні невеликих кількостей кислоти чи лугу)
Розчини, що містять слабку кислоту і сіль цієї кислоти з сильною основою (рН=4-7) |
Розчини, що містять слабку основу і сіль цієї основи з сильною кислотою (рН=7-10) |
Розчини, що містять солі багатоосновних слабких кислот рН=7,35-7,45 |
СН3СООNa → СН3СОО- + Na+ NaНСО3 → Na+ + НСО3- (дисоціація солей) Часткова дисоціація слабких кислот: СН3СООН +Н2О ↔ СН3СОО- + Н3О+ Н2СО3 + Н2О↔НСО3- + Н3О+
рівновага зміщується ліворуч, відновлюється рН. СН3СОО- + Н+↔СН3СООН
Н3О+ + НСО3-→Н2СО3 + Н2О (Надлишкова концентрація Н+ нейтралізується взаємодією з НСО3-). Надлишок Н2СО3 гідролізується у присутності ферменту карбоангідрази: Н2СО3↔ СО2 + Н2О СО2 вилучається через легені. |
NH4Cl→ NH4+ + Cl- (повна дисоціація солі). NH4OH ↔ NH4+ + ОН- (часткова дисоціація).
Н3О+ + NН3Н2О → 2Н2О + NН4+ (нейтралізація кислоти іонами ОН-, [ОН -]=const, рН=const
рівновага при дисоціації аміаку зміщується ліворуч, [ОН-]=const NH4+ + ОН-↔ NH4OH (ОН- зв’язуються з іонами NH4+, утвореними в результаті дисоціації солі, рН=const). |
Na2HPO4↔ 2Na+ + HPO4- NaH2PO4↔ Na+ + H2PO4- Між гідрофосфат- і дигідрофосфат-іонами встано-влюється рівновага: НРО42- + Н3О+ ↔Н2РО4- + Н2О НРО42- + Н2О ↔Н2РО4- + ОН-
НРО42- (аніон дуже слабкої кислоти, акцептор іонів Н3О+) зв’язує іони водню сильної кислоти з утворенням Н2РО4- НРО42- + Н3О+ →Н2РО4- + Н2О
Н2РО4- + ОН-→ НРО42- + Н2О |
Н3О+ + ОН- → 2Н2О Нейтралізація іонами Н3О+ гідроксид-іонів, рівновага в реакції дисоціації кислоти зміщується праворуч, рН-const СН3СООН + NaОН → СН3СООNa + Н2О. При надходженні лужних продуктів – кисла частина буферної системи нейтралізує луг: Н2СО3 + ОН-→НСО3- + Н2О НСО3- ‑ виводиться нирками. Висновки:
рН=рКа
+ lg |
Висновки:
рН=14
– рКв
+ lg |
Висновки:
НРО42- зв’язують іони Н+ (рівновага зміщується праворуч, утворюються Н2РО4-);
Н2РО4- зв’язують іони ОН- (рівновага зміщується ліворуч, утворюються НРО42-). рН
= рКн2ро4-
‑ lg |
Таким чином, рН буферного розчину певного складу визначається відношенням концентрації кислоти і солі або основи і солі (не залежить від розведення).
При зміні об’єму розчину концентрація кожного компоненту змінюється в однакове число раз і їх співвідношення залишається сталим.
Буферна суміш підтримує сталим значення рН за умови: кількість доданих до розчину кислоти або основи не перевищує граничної величини (буферна ємність).
Здатність буферних розчинів протидіяти зміні рН – буферна дія.
Межі, в яких проявляється буферна дія, називається буферна ємність (кількість моль еквівалентів сильної кислоти або сильної основи, що треба додати до 1л буферного розчину для зміни рН середовища на одиницю).
Буферна ємність залежить від:
природи і концентрації компонентів буферного розчину;
співвідношення концентрацій компонентів.
Ч
калібрування
рН-метрів; визначення
твердості води; підтримання
сталості рН культурних середовищ для
вирощування бактерій.В хімічному аналізі і мікробіологічних дослідженнях:
им більша концентрація компонентів,
чим ближче співвідношення концентрацій:
кислота – сіль, основа – сіль до одиниці,
тим більша буферна ємність.
В
технологічних процесах: електрохімічне
нанесення захисних шарів; виробництво
барвників; виробництво
шкіри; виробництво
фотоматеріалів.
Роль буферних систем



В
життєдіяльності живих організмів: підтримання
сталості КОС (кислотно-основного стану)
для оптимального функціонування
організму. рН крові в нормі = 7,35 – 7,45
(слабколужне середовище). рН<7,35
– ацидоз; рН>7,45
– алкалоз. Підтримка
КОС у межах, оптимальних для життєдіяльності,
забезпечується: біохімічними
буферними системами (гідрокарбонатна,
фосфатна, гемоглобінові, білкова); фізіологічними
буферними системами (легені, нирки,
шлунково-кишковий тракт); фізичними
буферними системами (розбавлення
позасудинною рідиною кислих і лужних
продуктів, що надходять у кров –
зменшення їх концентрацій).
Типи реакцій гідролізу |
||
Тип реакції гідролізу |
Хімічне рівняння гідролізу |
Вираз константи гідролізу |
Гідроліз солі, яка при дисоціації у водному розчині утворює катіон сферичної конфігурації і аніон – сильний електродонор (аніон слабкої кислоти – гідроліз за аніоном): |
СН3СООNa ↔ Na+ + СН3СОО- СН3СОО- + НОН ↔ СН3СООН + ОН- СН3СООNa + Н2О ↔ СН3СООН + NaОН |
Кг
=
рН > 7, середовище лужне. |
Гідроліз солі, яка при дисоціації у водному розчині утворює катіон – сильний комплексоутворювач і аніон - слабкий електродонор (катіон слабкої основи – гідроліз за катіоном): |
NH4Cl ↔ NH4+ + Cl- NH4+ + HOH ↔ NH4OH + H+ NH4Cl + H2O ↔ NH4OH + HCl |
Кг
= рН < 7, середовище кисле. |
Гідроліз солі, яка при дисоціації у водному розчині утворює катіон сильний комплексоутворювач і аніон – сильний електродонор (катіон слабкої основи і аніон слабкої кислоти – гідроліз солі за катіоном і аніоном): |
СН3СООNH4 ↔ СН3СОО- + NH4+ CH3COO- + NH4+ + НОН ↔ СН3СООН + NH4+ОН |
Кг
=
|
Якщо рКкисл. = рКосн.(рН=7), реакція середовища нейтральна; рКкисл. > рКосн. (рН>7), реакція середовища лужна; рКкисл. < рКосн. (рН<7), реакція середовища кисла. |
||
 =
=
 =
=
 =
=

Використання реакцій гідролізу в якісному аналізі:
як характерні реакції відкриття катіонів Fe3+, Sb3+, Br3+ і аніонів СН3СОО-, SіО32-,
для розділення Cr3+ і Al3+ - іонів;
для регулювання рН і рОН розчинів (додавання NH4+, СН3СОО-) тощо.
У практиці якісного аналізу найчастіше мають справу з гідролізом солей і солеподібних сполук.
Кількісні характеристики гідролізу


Константа
гідролізу
(Кг) (відношення
добутку молярних концентрацій речовин
продуктів гідролізу до молярної
концентрації речовин негідролізованих
іонів солі у розчині).
Записують
скорочене іонно-молекулярне рівняння
реакції гідролізу. Кг
=
![]()
Ступінь
гідролізу
(h)
(відношення
кількості речовини солі, що гідролізувала
до загальної кількості речовини Х). h
=
Одиниці
вимірювання:
в частках одиниці або у відсотках.![]()

Шляхи посилення гідролізу:
розведення розчинів;
підвищення температури;
видалення продуктів гідролізу;
додавання до розчину: катіонів – сильних комплексоутворювачів і аніонів – сильних донорів пар електронів.
Лекція №6. Рівновага в гетерогенній системі
Основні питання:
Гетерогенна система. Рівновага в гетерогенній системі.
Добуток розчинності.
Умови утворення і випадання осаду при проведенні аналітичних реакцій.
Використання правила ДР в аналітичній хімії.
Гетерогенна система – це система, яка складається з кількох фаз. Реакції, що відбуваються в гетерогенній системі, називаються гетерогенними реакціями.
Приклади: осадження AgCl, BaSO4 тощо.
Рівновага в гетерогенній (неоднорідній) системі: Осад ↔ Розчин.
В результаті взаємно протилежних процесів – осадження і розчинення – між осадом і розчиненою частиною речовини встановлюється динамічна рівновага, при якій швидкість розчинення υ1 дорівнює швидкості осадження υ2.
Дисоціація важкорозчинного електроліту: АаВb↔ аА+ + bВ –
Згідно
закону дії мас: Крівн.=
![]() ,
або
,
або
[А+]а · [В-]b = К(АаВb) · [АаВb] = const
Добуток молярних концентрацій іонів малорозчинного електроліту в його насиченому водному розчині за сталої температури і тиску, називають добутком розчинності.
Позначають ДР з індексом електроліту:
ДР (АаВb) = [А+]а · [В-]b = const (константа добутку розчинності речовини АаВb)
[А+] і [В -] – рівноважні концентрації катіонів і аніонів, утворених в результаті електролітичної дисоціації електроліту АаВb; (моль/дм3, моль/л);
а, b – число атомів або атомних груп в молекулі. Значення ДР вказані в довідниках.
Фізична сутність рівняння ДР: в насиченому водному розчині малорозчинного електроліту в стані рівноваги за даних температури та тиску величина добутку розчинності є сталою величиною, незалежно від змін концентрацій окремих іонів.
Дозволяє враховувати зміни концентрацій одних іонів малорозчинного електроліту залежно від змін концентрації інших.
ДР залежить від:
температури;
іонної сили розчину;
коефіцієнта активності іонів.
Величиною ДР користуються лише по відношенню до електролітів, розчинність яких у воді не більша, ніж
1·10-2моль/дм3.
Якщо електроліти мають однотипну формулу, то чим менше значення ДР, тим менша розчинність малорозчинного електроліту.
Напрям реакції обміну між двома електролітами у водному розчині визначається можливістю утворення між іонами малорозчинної сполуки (осаду).
Застосування правила добутку розчинності в аналітичній хімії
для вирішення питань утворення і розчинення осадів;
Утворення або розчинення осаду – аналітичний ефект, який фіксується органолептично (візуально).
Утворення осаду – результат зниження енергії Гіббса в системі, яке обумовлене:
зміною характеру і міцності хімічних зв’язків у вихідних і кінцевих продуктах системи (ентальпійний фактор ΔН<0);
невпорядкованістю системи при зливанні двох водних розчинів, які містять гідратовані іони, пов’язане з природою іонів і природою розчинника (ентропійний фактор).
Фактор утворення осаду – тверда фаза більш упорядкована, ентропія – менша.
Фактор посилення впорядкованості розчинника.
для обчислення розчинності малорозчинних сполук;
Розчинність малорозчинного електроліту типу АаВb обчислюють за формулою: (кількість молей речовини, що міститься в 1л насиченого розчину за даної температури):
S(АаВb)
=
![]() ,
[S]
‑
[моль/л]
,
[S]
‑
[моль/л]
для вирішення питань послідовності утворення і випадання осадів (дія групових реактивів).
Дробне осадження – це метод, за допомогою якого одним і тим же осаджувачем послідовно розділяють декілька іонів, користуючись різними величинами добутку розчинності утворених сполук. В якісному аналізу використовують у тих випадках, коли осади відрізняються за зовнішнім виглядом (забарвлення). Сутність: спочатку випадає в осад сполука, добуток розчинності якої має меншу величину.
Умови утворення і випадання осаду
Осад малорозчинного електроліту утворюється тоді, коли після змішування розчинів реагентів добуток молярних концентрацій речовин катіонів і аніонів буде більшим, ніж ДР осаду за даної температури (ЙД > ДР осаду):
[А+]а · [В-]b > ДР(АаВb) – утворення осаду (пересичений розчин, переважає процес осадження, процес розчинення термодинамічно неможливий). При невеликому перевищенні ДР розчин стає пересиченим, але осад деякий час не випадає.
[А+]а · [В-]b = ДР(АаВb) – осад не випадає (насичений розчин, динамічна рівновага).
[А+]а · [В-]b < ДР(АаВb) – розчинення осаду (ненасичений розчин)
Осадження можна вважати практично повним, якщо в розчині залишається така кількість речовини осаджуваних іонів, яка не заважає в подальших операціях розділення і виявлення іонів. (ДР ≠ 0, осадження ніколи не буває повним).
Для більш повного осадження до розчину додають надлишок реагенту – осаджувача, проте дуже великий надлишок осаджувача викликає розчинення осаду (сольовий ефект).
Вплив електролітів на розчинність осадів
Сольовий ефект – це підвищення розчинності малорозчинних електролітів, які перебувають у рівновазі з осадом, при додаванні до них сильних електролітів, що не містять однойменних іонів з осадом.
Розчинність малорозчинного електроліту у присутності іншого електроліту, що не містить з ним однойменних іонів збільшується, що пояснюється зростанням іонної сили розчину і зменшенням коефіцієнту активності кожного з іонів.
Для важко розчинного електроліту:
ДР(АаВb) = [А+]а · [В-]b∙f(А+) ∙f (B-), де
f(А+) ∙f (B-) – коефіцієнти активності іонів.
Так як добуток розчинності – величина стала, тому із збільшенням іонної сили розчину концентрація іонів малорозчинного електроліту збільшується, тобто збільшується розчинність осаду.
Приклад1. Обчислити розчинність АgCI в 0,1М розчині КNО3.
Позначивши розчинність АgCI в 0,1М розчині КNО3 через х моль/л, коефіцієнт активності однозарядного іона через f, одержимо:
ДР
(АgCI) = х2∙
f2
![]()
Коефіцієнт активності однозарядного іона в розчині КNО3 з молярною концентрацією калій нітрату (V) 0,1 моль/л, дорівнює 0,76. Звідси, розчинність АgCI в 0,1М розчині КNО3:
![]()
Розчинність
АgCI
в чистій воді складає 1,05∙10-5моль/л.
Відповідно розчинність АgCI
в 0,1М розчині КNО3
в 1,3 рази більша, ніж в чистій воді (![]() )
)
Вплив однойменних іонів:
при додаванні однойменних іонів до насичених розчинів малорозчинних електролітів розчинність цих малорозчинних електролітів зменшується.
Розглянемо розчинність кальцій оксалату СаС2О4 у присутності (NH4)2С2О4.
ДР(СаС2О4) = а (Са2+) ∙ а (С2О42-).
Активність оксалат-іону в розчині є сумою величин активності оксалат-іону, одержаного внаслідок незначного розчинення осаду та активності оксалат-іону з розчинного амоній оксалату.
Звідси, величина активності а (С2О42-) у присутності (NH4)2С2О4 > а (С2О42-) в насиченому розчині СаС2О4. Так як ДР(СаС2О4) = соnst (за даної температури), то величина а (Са2+) відповідно зменшиться, що спричиняє зменшення концентрації іонів кальцію в розчині (зменшення розчинності СаС2О4). Тому, розчинність СаС2О4 у присутності надлишку (NH4)2С2О4 (оксалат-іонів С2О42-) менша, ніж в чистій воді.
[А+]а · [В-]b > ДР(АаВb), розчин стає пересиченим, нестійким: при стоянні виділяє частину розчиненої речовини у вигляді осаду.
Розчинність осадів залежить від:
Концентрації іонів гідрогену (розчинність осадів в кислотах).
Дисоціація важкорозчинної сполуки: ВаСО3 Ва2+ + СО32-
При дії на осад ВаСО3 кислоти (іонів Н+) рівновага між осадом і розчином зміщується праворуч (згідно принципу Ле Шательє) тому, що карбонат-іон зв’язується з іонами гідрогену, утворюючи слабку карбонатну кислоту:
2Н+ + СО32- = Н2СО3,
яка розкладається з виділенням СО2; відповідно зменшується концентрація карбонат-іонів СО32-. Як результат – збільшення розчинності осаду ВаСО3.
Дія кислоти на осад залежить від:
Значення добутку розчинності важкорозчинної солі (чим більше ДРсолі, тим більша розчинність осаду цієї солі);
Величини константи дисоціації утвореної слабкої кислоти (чим менше Кдис. слабкої кислоти, тим більша розчинність осаду).
Температури.
Якщо розчинність речовини у воді супроводжується поглинанням теплоти, то підвищення температури спричиняє збільшення розчинності осаду (принцип Ле Шательє) – осадження необхідно проводити на холоді.
Наприклад, розчинність РbCI2 за кімнатної температури дорівнює 10,9г/л, а при 100 С – 333,8г/л.
Природи розчинника.
При додаванні до води органічних розчинників розчинність неорганічних солей, як правило, зменшується тому, що розчинність полярних речовин у полярних розчинниках більша, ніж у неполярних.
Висновки:
ДР – іонний добуток, що відповідає насиченому водному розчину при даній температурі.
У ненасиченому розчині (υ1 > υ2): [А+]а · [В-]b < ДР(АаВb), тому осад розчинятиметься доти іонний добуток не досягне величини добутку розчинності.
У пересиченому розчині (υ1 < υ2): [А+]а · [В-]b > ДР(АаВb), тому осад утворюватиметься доти іонний добуток не досягне величини добутку розчинності.
Розчинність малорозчинного електроліту типу АаВb обчислюють за формулою: (кількість молей речовини, що міститься в 1л насиченого розчину за даної температури):
S(АаВb) = , [S] ‑ [моль/л]
Вплив електролітів на розчинність осадів:
Розчинність малорозчинної речовини у присутності інших електролітів, які мають з ним спільні іони, менша розчинності в чистій воді;
Розчинність малорозчинного осаду у воді збільшується при додаванні до нього розчину сильного електроліту, що не містить спільного іону з осадом (сольовий ефект).
Чим менша тенденція до переходу осаду в розчин, тим менше добуток розчинності і розчинність відповідної сполуки;
Напрям реакції обміну між двома електролітами у розчині визначається можливістю утворення між іонами малорозчинної сполуки, яка випадає в осад (чим менша розчинність утвореної малорозчинної сполуки, тим сильніше рівновага зміщується в сторону її утворення).
Лекція №7. Системи якісного аналізу катіонів
Основні питання:
Системи якісного аналізу катіонів: сульфідна, кислотно-лужна.
Систематичний і дробний аналіз.
Схема загальної характеристики аналітичної групи катіонів.

Сульфідна система якісного аналізу катіонів
Аналітична група |
Груповий реактив |
Катіони |
І аналітична група |
Немає групового реактиву |
K+, Na+, NH4+, (Mg2+) (безбарвні, солі добре розчинні у воді, солі NH4+ розкладаються при нагріванні ). |
ІІ аналітична група |
(NH4)2CO3 у присутності NH4OH і NH4Cl |
(Mg2+), Ca2+, Ba2+ |
ІІІ аналітична група |
(NH4)2S у присутності NH4OH |
Al3+, Cr3+, Fe2+, Fe3+, Mn2+, Zn2+ |
ІV аналітична група |
H2S у присутності HCl |
Ag+, Pb2+,Cu2+ |
Кислотно-лужна система якісного аналізу катіонів
№ групи |
Катіони |
Груповий реактив |
1. |
Na+, K+, NH4+ |
Відсутній |
2. |
Ag+, Hg22+, Pb2+ |
HCl (розведена) |
3. |
Ca2+, Sr2+, Ba2+ |
H2SO4 (розведена) |
4. |
Al3+, Cr3+, Zn2+, As3+, As(V), Sn2+, Sn(IV) |
КОН або NaOH (надлишок) |
5. |
Fe2+, Fe3+, Mg2+, Mn2+, Bi3+, Sb3+, Sb(V) |
КОН або NaOH |
6. |
Cu2+, Co2+, Cd2+, Hg2+, Ni2+ |
NH3·H2O (надлишок) |
Кислотно – лужна система якісного аналізу свідчить, що аналітичні групи катіонів у більшості відповідають
групам періодичної системи хімічних елементів Д.І. Менделєєва.
Переваги кислотно – лужної класифікації катіонів:
Аналітичні групи катіонів цієї системи близькі до природних груп хімічних елементів у періодичній системі хімічних елементів Д. І. Менделєєва;
Теоретичні основи кислотно – лужної класифікації найбільше пов’язані з неорганічною хімією;
Для практичного використання потрібно витратити менше часу;
Не використовуються шкідливі для здоров`я сірководень та сульфід амонію.
Основні етапи якісного аналізу
Відбір проби і підготовка її до аналізу;
Попередні дослідження;
Вибір розчинника;
Одержання розчину зразка;
Проведення аналізу вибраним методом.
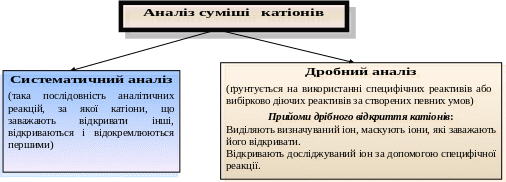
Схема загальної характеристики аналітичної групи катіонів
Перелік іонів, що входять до складу аналітичної групи.
Груповий реактив на катіони даної аналітичної групи.
Ефективна валентність атома, ступінь окислення атома, заряд його іона, забарвлення іонів в розчинах.
Хімічний характер оксидів, гідроксидів, їх кислотно – основні властивості.
Розчинність солей даних іонів у воді.
Гідроліз солей.
Окислювально – відновні властивості іонів.
Здатність до комплексоутворення.
Лекція №8. Катіони і аналітичної групи. Аналіз суміші катіонів
Основні питання:
Загальна характеристика І аналітичної групи катіонів.
Біохімічна роль катіонів І аналітичної групи.
Якісні реакції катіонів І аналітичної групи.
Аналіз суміші катіонів І аналітичної групи.
Загальна характеристика катіонів і аналітичної групи
До катіонів І аналітичної групи належать катіони Na+, К+, NH4+. Загального реактиву, який осаджував би всі катіони І аналітичної групи, немає, оскільки майже всі сполуки катіонів калію, натрію, амонію розчинні у воді.
Катіони Na+, К+ утворюють хімічні елементи І групи головної підгрупи Періодичної системи хімічних елементів: S-елементи, типові елементи–метали, атоми мають великий радіус і електронну конфігурацію зовнішнього рівня nS1. Характерний ступінь окислення ‑ „+1”, заряд іонів ‑ „1+”. Не характерні реакції комплексоутворення.
Оксиди Na2O, K2O виявляють основний характер, добре розчинні у воді, водні розчини їх ‑ розчинні основи (луги): NаОН, КОН. Гідроксиди калію, натрію – білі, тверді, гігроскопічні, дуже їдкі, легкоплавкі маси. У розплавленому стані сильно діють на скляний, фарфоровий, платиновий посуд (у присутності кисню) – плавлення лугів проводять у срібних тиглях.
Солі Na+, К+ - іонні сполуки, більшість яких добре розчинні у воді, у розчинах – безбарвні. Водні розчини солей калію, натрію мають нейтральну реакцію середовища ( з аніонами сильних кислот), або лужну (з аніонами слабких кислот) – результат гідролізу солі за аніоном.
NH4+ ‑ катіон має радіус близький до радіуса К+‑ катіона, безбарвний в розчині, відновник, комплексоутворювач. Водний розчин гідроксиду амонію NH3∙H2O – слабка основа, має слабко лужну реакцію середовища. Солі амонію добре розчинні у воді, внаслідок гідролізу мають кислу (з аніоном сильної кислоти), слабко лужну або нейтральну реакцію середовища (з аніонами слабких кислот). Солі амонію ‑ леткі за звичайних умов, розкладаються при нагріванні чи прожарюванні.
Перелік продуктів, багатих на катіони І аналітичної групи |
|
Найменування речовин |
Основні джерела |
Na |
Хліб (крім безсольового), масло вершкове, сири, особливо розсільні; ковбасні вироби, консерви м’ясні, рибні, кухонна сіль, питна сода |
K |
Картопля, салат, гарбуз, кабачки, зелений горошок, буряк; вівсяна крупа; сухофрукти, особливо ізюм, курага. |
NH4+ |
Утворюється під час мікробіологічного розкладу білків в продуктах харчування |
Біохімічна роль катіонів і аналітичної групи
Катіон Na+ міститься в міжклітинному просторі, вміст в організмі – 0,15%. Добова потреба – 10-15 г (у вигляді NaCl).
Роль Na+:
Підтримання сталості осмотичного тиску та об’єму фізичних рідин організму;
Сприяння затримці води в організмі;
Участь в транспорті амінокислот, цукрів та інших сполук;
Участь в передачі нервових імпульсів, вплив на збудливість м’язових волокон та серцево-судинної системи.
Нестача Na+: втрата апетиту, апатія, тахікардія.
Надлишок Na+: порушення діяльності серцево-судинної системи, нирок, підвищення тиску, поява набряків,
судом, порушення виведення шлаків з організму.
Катіон К+ міститься у всіх тканинах організму і травних соках, вміст в організмі – 0,35%. Добова потреба – 2-3 г.
Роль К+:
Передача нервових імпульсів у м’язових і нервових тканинах;
Забезпечення осмотичних явищ;
Стимулювання діяльності серцевих м’язів.
Нестача К+: порушення синтезу глікогену, поява серцевої аритмії, посилення розщеплення білків.
Надлишок К+: порушення розумової діяльності, фізичного розвитку, функціонування систем організму, посилення виділення натрію з організму.
Якісні реакції катіонів І аналітичної групи
Катіон |
Характерні реакції та умови їх проведення |
Na+ |
NaCl + K[Sb(OH)6] ® Na[Sb(OH)6]↓ + KCl Na+ + [Sb(OH)6]- ® Na[Sb(OH)6]↓ Умови: рН=7,5-8, концентрований розчин, на холоді, наявність центрів кристалізації, відсутність NH4+ та Mg2+ |
|
|
К+ |
2KNO3 + Na3[Cо(NO2)6] ® K2Na[Cо(NO2)6]↓ + 2NaNO3 2K+ + Na+ + [Cо(NO2)6]3- ® K2Na[Cо(NO2)6]↓ Умови: нейтральне або оцтовокисле середовище (рН=4-7), заважають NH4+, на холоді |
КСl + NаНС4Н4О6 → KНС4Н4О6↓ + NaCl К+ + НС4Н4О6- → KНС4Н4О6↓ Умови: рН = 7, наявність центрів кристалізації, на холоді, відсутність Са2+,Ва2+,NН4+ - катіонів , окисників (для запобігання окисленню тартратної кислоти) |
|
Умови: слід розглядати полум’я через сине скло (заважають Na+) |
|
NH4+ |
NH4Cl + KOH ® KCl + NH3↑ + H2O NH4+ + OH- ® NH3↑ + H2O |
NH4+ + 2[HgI4]2- + 2OH- ® NH2Hg2I3↓ + 5I- + 2H2O В контролі якості м’яса для визначення наявності аміаку (мікробіологічний процес розщеплення білків) |
|
(NH4)2CO3 ® 2NH3 + CO2↑ + H2O |
Аналіз суміші катіонів І аналітичної групи
Перевірка на наявність катіона NH4+: |
Досліджуваний розчин + реактив Несслера червоно-бурий осад (жовто-буре забарвлення) |

Відокремлення катіона NH4+: |
Досліджуваний розчин випаровують у фарфоровій чашці до припинення виділення білого диму. Чашку охолоджують. Сухий залишок подрібнюють скляною паличкою, додають концентровану HNO3. Розчин випаровують. Залишок прожарюють. |
Перевірка на повноту видалення солей NH4+: |
Реактив Несслера + кристал сухого залишку відсутність червоно-бурого осаду або бурого забарвлення розчину |
Відкриття катіона К+ (після повного видалення солей NH4+): Проводити в пробірці! |
Сухий залишок + 1-2 мл Н2О (дистильована) Розчин (І) Розчин (І) + 1-2 крап. фенолфталеїну безбарвний розчин + 2н. розчин NaOH поява рожевого забарвлення (рН>8) → Розчин (ІІ) Розчин (ІІ) + 2н. розчин CH3COOH до знебарвлення (рН = 3-4) Розчин (ІІІ) Розчин (ІІІ) + 3-5 крапель Na3 Cо(NO2)6 осад жовтого кольору |
Відкриття катіона Na+ (при відсутності NH4+, Mg2+): Проводити в пробірці! |
Розчин (ІІ) + K[Sb(OH)6] осад білого кольору |
Лекція № 9. Катіони іі аналітичної групи. Аналіз суміші катіонів
Основні питання:
Загальна характеристика катіонів ІІ аналітичної групи.
Характеристика групового реактиву.
Якісні реакції катіонів ІІ аналітичної групи.
Аналіз суміші катіонів.
Загальна характеристика катіонів іі аналітичної групи
До катіонів другої аналітичної групи належать катіони Са2+, Ва2+, Мg2+ (знаходяться в ІІ групі головній підгрупі Періодичної системи хімічних елементів Д.І.Менделєєва). Груповим реактивом на катіони даної групи є (NH4)2CO3 амоній карбонат у присутності NH4Cl і NH4OH.
Належать до S-елементів, типові елементи – метали, сильні відновники атоми та прості речовини). Загальна електронна формула зовнішнього електронного рівня nS2. Характерний ступінь окислення „+2”, заряд іонів „2+”. Катіони Са2+, Мg2+, Ва2+ стійкі до відновлення та окислення в розчинах.
Оксиди МgО, СаО, ВаО ‑ стійкі, міцні, тугоплавкі, гігроскопічні, білі сполуки. Оксиди кальцію та барію енергійно взаємодіють з водою з виділенням великої кількості теплоти, утворюючи гідроксиди сильні основи. Ca(OH)2 – досить сильна основа, Ba(OH)2 – луг, Mg(OH)2 – малорозчинна сполука.
Солі катіонів Мg2+, Са2+, Ва2+ ‑ іонні сполуки (іонний характер обумовлений збільшенням атомного радіуса і зменшенням іонного потенціалу). Більшість сполук катіонів кальцію, магнію, барію – безбарвні в розчинах (забарвлення обумовлене лише забарвленням аніонів, що входять до складу їх сполук). У водних розчинах існують у вигляді безбарвних аквакомплексів [Ме(OH2)6]2+.
Добре розчинні у воді – нітрати, хлориди, броміди. Фосфати, сульфіти, карбонати, оксалати – практично нерозчинні у воді, але розчинні в сильних кислотах. MgSO4 – добре розчиняється у воді, CaSO4 – малорозчинний, BaSO4 – не розчиняється у воді та кислотах. Кислі солі – добре розчинні у воді.
Специфічних реакцій на окремі катіони ІІ аналітичної групи немає, тому виявляють катіони Мg2+, Са2+, Ва2+ за допомогою загально аналітичних реакцій.
Перелік харчових продуктів, багатих на катіони ІІ аналітичної групи
Назва речовини |
Основні джерела |
Са |
Молоко та молочні продукти; петрушка – зелень; кріп і цибуля зелена; хурма японська; сухофрукти. |
Мg |
Крупи – пшоно, вівсянка, гречана, ячна, перлова; житній хліб, сухофрукти. |
Антихарчові речовини та шляхи усунення їх впливу
Інгібіторна харчова речовина |
Природний антихарчовий фактор |
Джерела та умови дії |
Шляхи усунення впливу |
Мінеральні речовини: Са, Мg, Мп |
Щавлева кислота |
Щавель, шпинат, ревінь, інжир, горниця, картопля – при надлишковому вживанні |
Збільшення вживання джерел засвоюваного кальцію та інших катіонів |
Бобові, деякі крупи, висівки – при недостатній тепловій обробці. |
Теплова обробка |
||
Са, Мg |
Фітин |
Чорний хліб – при надлишковому вживанні. |
Вживання в межах рекомендованої норми. |
Са, Мg, Na |
Кофеїн |
Кава – при надлишковому вживанні. |
Помірне вживання |
Са |
Надлишок фосфору |
Більшість продуктів масового вживання |
Щоденне вживання молока або молочних продуктів, сиру |
Біохімічна роль катіонів іі аналітичної групи
Са2+ міститься у кістках у вигляді нерозчинних солей фосфорної кислоти Н3РО4 – гідроксилапатитів, решта – в іонному вигляді, в комплексі з білками альбумінової фракції в усіх тканинах і рідинах організму (в крові у вигляді СаСl2).
Вміст в організмі – 1,5%, добова потреба – 0,9-1 г.
Мg2+ та Са2+ виявляють антагоністичну дію: при надлишку іонів магнію пригнічується всмоктування іонів кальцію і заміна його в складі органічних сполук на магній..
Роль Са2+:
участь в процесах скорочення м’язів;
участь в процесах зсідання крові;
формування опірних і покривних тканин.
Мg2+ міститься в плазмі крові, органах, тканинах, у вигляді фосфатів входить до складу кісткової тканини. Вміст в організмі – 0,15%, добова потреба 0,6-0,7 г.
Роль Мg2+:
забезпечення формування просторової конформації білків і нуклеїнових кислот;
підтримання структури клітинних органел (мітохондрій, рибосом), що необхідно для виконання ними біологічних функцій (здатність іонів Мg2+ до комплексоутворення).
забезпечення процесів трансляції, транскрипції та реплікації;
у ферментативних процесах обміну білків, вуглеводів;
сприяння підвищенню стійкості організму проти інфекційних захворювань.
Нестача Мg2+ спричиняє апатію, слабкість, сонливість, кальцифікацію артеріальних судин, серцевого м’яза, нирок.
Дія групового реактиву на катіони іі аналітичної групи
(NH4)2CO3 – амоній карбонат (карбонат амонію) у присутності NH4Cl і NH4OH при нагріванні осаджує катіони Са2+ та Ва2+, а катіони Мg2+ залишаються в розчині.
Гідроліз групового реактиву карбонату амонію:
(NH4)2CO3 + Н2О ↔ NH4OH + NH4HCO3
(збільшення концентрації NH4OH сприяє зміщенню рівноваги в бік зворотної реакції – спричиняє зменшення ступеня гідролізу групового реактиву).
2NH4HCO3 + CaCl2 Ca(HCO3)2 + 2NH4Cl (утворюються розчинні у воді гідрогенкарбонати кальцію та барію)
2NH4HCO3 + BaCl2 Ba(HCO3)2 + 2NH4Cl
Роль NH4OH:
запобігання гідролізу групового реактиву(NH4)2CO3;
запобігання утворенню розчинних гідрокарбонатів Са2+ та Ва2+.
Роль нагрівання:
кислі солі розкладаються з утворенням карбонатів: Ca(HCO3)2 → СаСО3 + Н2О + СО2;
аморфні осади переходять в кристалічні;
Роль NH4Cl: сприяє розчиненню основних карбонатів Мg2+ і переходу їх у розчин.
2МgCl2 + 2(NH4)2CO3 + Н2О (МgОН)2CO3↓ + 4NH4Cl + СО2↑
2Мg2+ + 2CO32- + Н2О (МgОН)2CO3↓ + СО2↑;
(МgОН)2CO3↓ + 4NH4Cl = 2МgCl2 + 2NH4OH + (NH4)2CO3
(МgОН)2CO3↓ + 2NH4+ = 2Мg2+ + 2NH4OH + CO32-
Характерні реакції катіонів ІІ аналітичної групи
Катіон |
Характерна реакція та умови її проведення |
Са2+ |
CaCl2 + (NH4)2CO3 → СаCO3↓ + 2NH4Cl Ca2+ + CO32- → СаCO3↓ Осад розчиняється в мінеральних кислотах і оцтовій кислоті: СаCO3↓ + 2НCl ® СаCl2 + Н2О + СО2↑ СаCO3↓ + 2Н+ ® Са2+ + Н2О + СО2↑ Умови: рН=7,5-8, середовище аміачне. |
СаCl2 + (NH4)2C2O4 ® CaC2O4↓ + 2NH4Cl Са2+ + C2O42- ® CaC2O4↓ Осад розчиняється в мінеральних кислотах, крім оцтової кислоти. Умови: рН=7-8, на холоді, заважають іони Ва2+ (утворюють аналогічний осад). |
|
На предметне скельце: 1 крап. СаСl2 + 1 крап. 1М Н2SO4→ нагрівають на спиртівці до появи білої кайми → утворення голчастих кристалів у вигляді пучків або зірочок (спостереження проводять під мікроскопом). |
|
|
|
Ва2+ |
ВаCl2 + (NH4)2CO3 ® ВаCO3↓ + 2NH4Cl Ва2+ +СO32- ® ВаCO3↓ Осад розчиняється в розведених мінеральних кислотах, крім Н2SО4: ВаCO3↓ + 2HNO3 ® Ba(NO3)2 + Н2О + СО2↑ ВаCO3↓ + 2H+ ® Ba2+ + Н2О + СО2↑ Умови: слабко лужне або аміачне середовище (NH4OH), рН=8 |
Катіон |
Характерна реакція та умови її проведення |
Ва2+ |
K2CrO4 + ВаCl2 ® BaCrO4↓ + KCl CrO42- + Ва2+ ® BaCrO4↓ Умови: нейтральне або оцтовокисле середовище. Осад розчиняється в мінеральних кислотах, крім сульфатної кислоти: BaCrO4↓ + 2HNO3 ® Ba(NO3)2 + Н2СrО4 BaCrO4↓ + 2H+ ® Ba2+ + Н2СrО4 |
Ва2+ |
|
ВаCl2 + Н2SO4 ® ВаSO4↓ + 2НCl Ва2+ + SO42- ® ВаSO4↓ Осад не розчиняється в кислотах і лугах, крім концентрованої сульфатної кислоти: ВаSO4↓ + Н2SO4 ® Ва(НSO4)2 (при нагріванні) ВаSO4↓ + 2Н+ + SO42- ® Ва2+ + 2НSO4- |
|
Мg2+ |
2(NH4)2CO3 + 2MgCl2 + Н2О = Mg2(OH)2CO3↓ + 4NH4Cl + CO2↑ Осад розчиняється в мінеральних кислотах, оцтовій кислоті, солях амонію: Mg2(OH)2CO3↓ + 4HCl ® 2MgCl2 + 3Н2О + СО2↑ Mg2(OH)2CO3↓ + 4H+ ® 2Mg2+ + 3Н2О + СО2↑ Mg2(OH)2CO3↓ + 4NH4Cl ® 2MgCl2 + (NH4)2CO3 +2NH4ОН |
MgCl2 + Na2HPO4 + NH4ОН ® MgNH4PO4↓ + Н2О + 2NaCl Mg2+ + HPO42- + NH4ОН ® MgNH4PO4↓ + Н2О Умови: (NH4Cl + NH4ОН) аміачне середовище, заважають солі Cа2+ та Ва2+ |
|
MgCl2 + 2КОН ® Mg(ОН)2↓ + 2КСl Умови: відсутність солей амонію. |
Аналіз суміші катіонів ІІ аналітичної групи
Попередні дослідження: |
(NH4)2CO3 + NH4ОН + NH4Cl → білий осад, що свідчить про наявність катіонів ІІ аналітичної групи. |

Відкриття катіонів Ва2+: |
Досліджуваний розчин + К2СrО4 + СН3СООН жовтий осад |
Відокремлення катіонів Ва2+: |
Досліджуваний розчин + К2СrО4 (надлишок) жовтий осад центрифугування перевірка на повноту осадження катіонів барію. |
Відкриття катіонів Са2+: |
Центрифугат І + (NH4)2C2O4 білий кристалічний осад |
Відокремлення катіонів Са2+: |
Центрифугат І + (NH4)2C2O4(надлишок) білий кристалічний осад центрифугування перевірка на повноту осадження катіонів кальцію. |
Відкриття катіонів Мg2+: |
Центрифугат ІІ + Na2HPO4 + NH4OH білий кристалічний осад |
Лекція №10. Катіони ііі аналітичної групи. Аналіз суміші катіонів
Основні питання:
1.Загальна характеристика катіонів ІІІ аналітичної групи.
2.Характеристика групового реактиву.
3.Загальні реакції катіонів ІІІ аналітичної групи.
4.Характерні реакції катіонів ІІІ аналітичної групи.
5.Аналіз суміші катіонів.
Загальна характеристика катіонів ііі аналітичної групи
До ІІІ аналітичної групи належать катіони Al3+, Zn2+, Fe3+, Cr3+, Mn2 +, які є хімічними елементами різних груп і підгруп Періодичної системи хімічних елементів. Катіон Al3+ належить до р-елементів, а катіони Zn2+, Fe3+, Cr3+, Mn2+ - до елементів вставних декад (d-елементів). Атоми р- і d-елементів, які утворюють дану групу, мають порівняно невеликі та близькі радіуси, тому є відновниками. Крім, катіонів цинку та алюмінію, виявляють змінні ступені окислення: „+2” (цинк, феррум, манган), „+3” (феррум, хром, алюміній), „+4” (манган), „+6” (хром), „+7”,”+6”(манган).
Катіони Al3+, Zn2+, Cr3+ утворені атомами типових амфотерних елементів (у атомах цих елементів р-підрівні не забудовані). Вакантні р- і d-атомні орбіталі здатні заповнюватися електронами за рахунок утворення ковалентних зв’язків за донорно – акцепторним механізмом (утворення стійких аквакомплексів і гідроксокомплексів). Координаційні числа центральних атомів - комплексоутворювачів – 4,6. Окремі катіони забарвлені у водних розчинах: іони Fе3+ мають жовто-коричневий колір, Mn2+ ‑ блідо – рожевий, Cr3+ ‑ темно-зелений.
Гідроксиди Al3+, Zn2+, Cr3+ виявляють амфотерні властивості. Гідроксиди Mn2+, Fe2+ виявляють основні властивості, нерозчинні у воді.
Катіони ІІІ аналітичної групи утворюють комплексні сполуки.
Розчинні солі – хлориди, сульфати, нітрати, кислі солі цих катіонів.
Практично нерозчинні солі – сульфіди, сульфіти, карбонати, силікати, фосфати, або не існують у водних розчинах (результат гідролізу).Більшість солей підлягає гідролізу тому, що утворені слабкими основами. Реакція середовища – кисла , якщо сіль утворена аніоном сильної кислоти.
Окремі катіони можна виявити за допомогою специфічних реакцій.
Біохімічна роль катіонів ііі аналітичної групи
Ферум (залізо) в організмі міститься 3-5г, 80% - в складі гемоглобіну, 5-10% - в складі міоглобіну, 1% - в складі дихальних ферментів (цитохроми). Резервне залізо – 20-25%.
Роль заліза:
участь в процесах транспорту і депонування кисню (гемоглобін міоглобін);
участь в процесах дихання (ферменти, що здійснюють транспорт електронів);
формування активних центрів окислювально-відновних ферментів;
участь в процесах кровотворення.
Нестача заліза: анемія, порушення апетиту, атрофічний гастрит, атонія скелетних м’язів, трофічна зміна нігтів.
Надлишок заліза: викликає порушення в роботі печінки, підшлункової залози, легень, серця.
Цинк в організмі міститься 1,5-2г; в м’язах, еритроцитах, плазмі, сперматозоїдах. Добова потреба – 10-15мг.
Роль цинку:
входить до складу металоферментів, що приймають участь в процесах обміну вуглеводів, жирів, синтезу білків і нуклеїнових кислот;
впливає на розвиток мозку, здійснення смакового сприймання.
Нестача цинку: порушення розвитку плоду новонароджених, дерматит, кровотеча, слабкість родової діяльності, знижується загоєння ран.
Надлишок цинку: викликає отруєння, проте його токсичність незначна, надлишок виводиться з організму.
Манган (марганець) міститься в організмі (10-20 мг) в клітинах, печінці, нирках, м’язах, мозку. Добова потреба – 3-6мг.
Роль марганцю:
позитивний вплив на процеси розвитку, росту, клітинного поділу, імунологічні процеси;
участь в процесах обміну вуглеводів (активатор ферментів), необхідний для нормальної секреції інсуліну;
участь в процесах обміну ліпідів (запобігає відкладанню жиру в печінці), синтезу холестерину;
позитивно впливає на синтез вітаміну С, підвищує біологічну активність вітамінів В2 і В12,
підсилює засвоєння йоду організмом та синтез гормонів щитовидної залози.
Нестача марганцю: діабет, судоми, анемія, послаблення імунітету, порушення ендокринних функцій центральної нервової системи.
Надлишок марганцю: порушення рухомої активності, психічні порушення.
Хром міститься в печінці, залозах внутрішньої секреції, кишках. Добова потреба 50-200мкг.
Роль хрому:
участь в процесах обміну вуглеводів, збільшує дію інсуліну (прискорює окислення глюкози, швидкість проникнення глюкози в клітини і перетворення її в жир, стимулює синтез глікогену);
участь в обміні ліпідів (знижує концентрацію тригліцеридів в плазмі крові);
участь в структурі та функціях нуклеїнових кислот.
Нестача хрому: збільшення концентрації інсуліну в крові, глюкозурія, гіперглікемія, затримання росту, збільшення концентрації холестерину в крові, порушення вищої нервової діяльності, зниження кількості сперматозоїдів.
Дія групового реактиву
Груповий реактив на катіони ІІ аналітичної групи - (NH4)2S (амоній сульфід) у присутності NH4OH.
(NH4)2S – сіль, утворена катіоном слабкої основи та аніоном слабкої кислоти, підлягає гідролізу:
(NH4)2S + H2O ↔ NH4HS + NH4OH І стадія
NH4HS + H2O ↔ NH4OH + H2S ІІ стадія
Утворені HS- гідросульфід – іони заважають повному осадженню катіонів ІІІ аналітичної групи тому, що гідросульфіди катіонів Al3+, Zn2+, Cr3+, Mn2+ добре розчинні у воді. Для зменшення ступеня гідролізу (NH4)2S добавляють NH4OH.
Груповий реактив при дії на суміш катіонів ІІІ аналітичної групи осаджує їх у вигляді сульфідів (сульфіди цинку, марганцю, заліза) або гідроксидів (гідроксиди алюмінію, хрому).
ДІЯ (NH4)2S СУЛЬФІДУ АМОНІЮ:
2AlCl3 + 3(NH4)2S + 6H2O → 2Al(OH)3↓ + 3H2S + 6NH4Cl
2Al3+ + 6Cl- + 6NH+4 + 3S2- + 6H2O → 2Al(OH)3↓ + 3H2S + 6NH4+ + 6Cl-
2Al3+ + 3S2- + 6H2O → 2Al(OH)3↓ + 3H2S↑
Aналогічно відбувається осадження Cr3+.
ZnCl2 + (NH4)2S → ZnS↓ + 2NH4Cl
Zn2+ + 2Cl- + 2NH4+ + S2- → ZnS↓ + 2NH4+ + 2Cl-
Zn2+ + S2- → ZnS↓
Aналогічно відбувається осадження Mn2+.
2Fe3+ + 6Cl- + 6NH4+ + 3S2- → Fe2S3 + 6NH4+ + 6Cl-
2Fe3+ + 3S2- → Fe2S3↓
При розчиненні осадів в HCl відбуваються реакції:
Al(OH)3↓ + 3HCl → AlCl3 + 3H2O
Al(OH)3↓ + 3H+ + 3Cl- → Аl3+ + 3Cl- + 3H2O
Al(OH)3↓ + 3H+ → Аl3+ + 3H2O
Аналогічно відбувається розчинення Cr(OH)3.
Сульфіди розчиняються кислоті згідно рівнянь:
ZnS↓ + 2HCl → ZnCl2 + H2S↑
ZnS↓ + 2H+ + 2Cl- → Zn2+ + 2Cl- + H2S↑
ZnS↓ + 2H+ → Zn2+ + H2S↑
Аналогічно відбувається розчинення MnS.
Fe2S3↓+ 6HCl → 2FeCl3 + 3H2S↑
Fe2S3↓+ 6H+ + 6Cl- → 2Fe3+ + 6Cl- + 3H2S↑
Fe2S3 ↓ + 6H+ → 2Fe3+ + 3H2S↑
ДІЯ NаOH В ЕКВІВАЛЕНТНІЙ КІЛЬКОСТІ:
AlCl3 + 3NaOH → 3NaCl + Al(OH)3↓
Al3+ + 3Cl- + 3Na+ + 3OH- → 3Na+ + 3Cl- + Al(OH)3↓
Al3+ + 3OH- → Al(OH)3↓
Аналогічно відбуваються реакції з Cr3+, Fe3+:
ZnCl2 + 2NaOH → Zn(OH)2 ↓ + 2NaCl
Zn2+ + 2Cl- + 2Na+ + 2OH- → Zn(OH)2 ↓ + 2Na+ + 2Cl-
Zn2+ + 2OH- → Zn(OH)2↓
Аналогічно відбувається реакція з Mn2+.
ДІЯ NаOH В НАДЛИШКУ:
Al(OH)3↓ + 3NaOH → Na[Al(OH)6];
Al(OH)3↓ + 3Na+ + 3OH- → 3Na+ + [Al(OH)6]3-
Al(OH)3↓ + 3OH- + [Al(OH)6]3-
Аналогічно відбувається реакція з Cr3+.
Zn(OH)2↓ + 2NaOH → Na2[Zn(OH)4]
Zn(OH)2↓ + 2Na+ + 2OH- → 2Na+ + [Zn(OH)4]2-
Zn(OH)2↓ + 2OH- → [Zn(OH)4]2-
ДІЯ НАДЛИШКУ Амоній ГІДРОКСИДУ NH4OH:
AlCl3 + 3NH4OH → Al(OH)3↓ + 3NH4Cl
Al3+ + 3Cl- + 3NH4OH → Al(OH)3↓ + 3NH4+ + 3Cl-
Al3+ + 3NH4OH → Al(OH)3↓ + 3NH4+
Аналогічно відбувається реакція з Cr3+, Fe3+.
ZnCl2 + 2NH4OH → Zn(OH)2↓ + 2NH4Cl
Zn2+ + 2Cl- + 2NH4OH → Zn(OH)2↓ + 2NH4+ + 2Cl-
Zn2+ + 2NH4OH → Zn(OH)2↓ + 2NH4-
Аналогічно відбувається реакція з Mn2+.
При дії надлишку NH4OH осад Zn(OH)2 розчиняється:
Zn(OH)2↓ + 4NH4OH → [Zn(NH3)4](OH)2 + 4H2O
гідроксид тетраамінцинку(ІІ)
ДІЯ Nа2HPO4 ГІДРОФОСФАТУ НАТРІЮ:
AlCl3 + 2Na2HPO4 → AlPO4 ↓ + 3NaCl + NaH2PO4
Al3+ + 3Cl- + 4Na+ + 2HPO42- → AlPO4 ↓ + 3Na+ + 3Cl- + Na+ +H2PO4-
Al3+ + 2HPO42- → AlPO4 ↓ + H2PO4-
Аналогічно відбувається реакція осадження фосфатів хрому(ІІІ), заліза(ІІІ).
3ZnCl2 + 4Na2HPO4 → Zn3(PO4)2 ↓ + 2NaH2PO4 + 6NaCl
3Zn2+ + 6Cl- + 8Na+ + 4HPO42- → Zn(PO4)2↓+ 2Na+ + 2H2PO4-↓ + 6Na+ + 6Cl-
3Zn2+ + 4HPO42- → Zn3(PO4)2↓ + 2H2PO4-
Аналогічно відбувається реакція з Mn2+.
Розчинення фосфатів в мінеральних кислотах:
AlPO4↓ + 3HCl → AlCl3 + H3PO4
AlPO4↓ + 3H+ + 3Cl- → Al3+ + 3Cl- + H3PO4
AlPO4↓ + 3H+ → Al3+ + H3PO4
Аналогічно відбувається реакція з CrPO4, FePO4
Zn3(PO4)2 ↓ + 6HCl → 3ZnCl2 + 2H3PO4
Zn3(PO4)2 ↓ + 6H+ + 6Cl- → 3Zn2+ + 6Cl-+ 2H3PO4
Zn3(PO4)2↓ + 6H+ → 3Zn2+ + 2H3PO4
Аналогічно розчиняється Mn3(PO4)2.
Розчинення фосфатів алюмінію, цинку, хрому в лугах:
AlPO4↓ + 4NaOH → NaAlO2 + Na3PO4 + 2H2O
AlPO4↓ + 4Na+ + 4OH- → Na+ +AlO2- + 3Na+ + PO43- + 2H20
AlPO4↓ + 4OH- → AlO2- + PO43- + 2H2O
ДІЯ НАТРІЙ КАРБОНАТУ Nа2CO3:
2AlCl3 + 3Na2CO3 + 3H2O → 2Al(OH)3↓ + 6NaCl + 3CO2↑
2Al3+ + 6Cl- +6Na+ + 3CO32- + 3H2O → 2Al(OH)3↓ + 6Na+ + 6Cl- + 3CO2↑
2Al3+ + 3CO32- + 3H2O → 2Al(OH)3↓ + 3CO2↑
Аналогічно відбуваються реакції з Сr3+ ;Fe3+
MnCl2 + Na2CO3 → MnCO3↓ + 2NaCl
Mn2+ + 2Cl- + 2Na+ +CO32- → MnCO3↓ + 2Na+ + 2Cl-
Mn2+ + CO32- → MnCO3 ↓
2ZnCl2 + 2Na2CO3 + H2O → Zn2(OH)2CO3↓ + 4NaCl + CO2↓
2Zn2+ + 4Cl- + 4Na+ + 2CO32- + H2O → Zn2(OH)2CO3↓ + CO2 ↑
Розчинення в кислотах:
Zn2(OH)2CO3↓ + 4HCl → 2ZnCl2 + CO2↑ + 3H2O
Zn2(OH)2CO3↓ + 4H+ + 4Cl- → 2Zn2+ + 4Cl- + CO2↑ + 3H2O
Zn2(OH)2CO3↓ + 4H+ → 2Zn2+ + CO2↑ +3H2O
Загальні реакції катіонів ІІІ аналітичної групи
Реактиви |
Катіони |
||||
Al3+ |
Zn2+ |
Cr3+ |
Fe3+ |
Mn2+ |
|
(NH4)2S |
Al(OH)3 білий осад |
ZnS білий осад |
Cr(OH)3 сіро-зелений осад |
Fe2S3 чорний осад |
MnS рожевий осад |
Всі осади розчиняються в 2н. розчині НСl |
|||||
NaOH в еквівалентній кількості |
Al(OH)3 білий осад |
Zn(OH)2 білий осад |
Cr(OH)3 сіро-зелений осад |
Fe(OH)3 червоно-бурий осад |
Mn(OH)2 білий осад MnO2 бурий осад |
NaOH в надлишку |
Na3[Al(OH)6] безбарвний розчин |
Na2[Zn(OH)4] безбарвний розчин |
Na3[Cr(OH)6] зелений розчин |
не розчиняються |
|
NH4OH в надлишку |
Al(OH)3 білий осад |
[Zn(NH3)4](OH)2 безбарвний розчин |
Cr(OH)3 сіро-зелений осад |
Fe(OH)3 червоно-бурий осад |
Mn(OH)2 білий осад буріє на повітрі MnO2 |
Na2HPO4 |
AlPO4 білий осад нерозчинний в CH3COOH, розчинний в лугах |
Zn3(PO4)2 білий осад розчинний в оцтовій кислоті, в розчинах лугів |
CrРO4 зелений осад нерозчинний в оцтовій кислоті розчинний в лугах |
FePO4 білий осад нерозчинний в оцтовій кислоті, лугах |
Mn3(PO4)2 білий осад розчинний в оцтовій кислоті, нерозчинний в лугах |
Всі осади розчинні в мінеральних кислотах |
|||||
Na2CO3 |
Al(OH)3 білий осад |
Zn2(OH)2CO3 білий осад |
Cr(OH)3 сіро-зелений осад |
Fe(OH)CO3 при кип’ятінні утворює Fe(OH)3 червоно-бурий |
MnCO3 білий осад |
Всі осади розчиняються в HCl та CH3COOH |
|||||
Характерні реакції катіонів ІІІ аналітичної групи
Катіон |
Характерна реакція та умови її проведення |
Fе3+ |
FeCl3 + 3KSCN → Fe(SCN)3 + 3KCl Fe3+ + 3SCN- → Fe(SCN)3 Умови: слабко кисле середовище. |
3K4[Fe(CN)6] + 4FeCl3 → 12KCl + Fe4([Fe(CN)6])3↓ гексаціаноферрат (ІІ)заліза(ІІІ) 3[Fe(CN)6]4- + 4Fe3+ → Fe4([Fe(CN)6])3↓ Умови: слабко кисле середовище. |
|
АІ3+ |
AlCl3+3NH4Cl+6KOH → Al(OH)3↓ + 3NH3↑ +6KCl + 3H2O Al3+ +3NH4+ +6OH- → Al(OH)3↓ + 3NH3↑ + 3H2O Умови: нагрівання до кипіння; лужне середовище; кристалічний NH4Cl. |
2Al2(SO4)3 + 2Co(NO3)2 → 2Co(AlO2)2 + 6SO3↑ + 4NO2↑ + O2↑ На смужку фільтрувального паперу послідовно наносять розчини солей алюмінію та кобальту, висушують, спалюють. Результат – зола синього кольору. Умови: заважають іони цинку, купруму, хрому, ніколу. |
|
Zn2+ |
ZnCl2 + H2S → ZnS↓ + 2HCl Zn2+ + S2- → ZnS↓ Умови (інші катіони ІІІ аналітичної групи за таких умов не осаджуються):
|
Катіон |
Характерна реакцій та умови її проведення |
Zn2+ |
На смужку фільтрувального паперу наносять 1-2 краплі водного розчину сульфату цинку та 1-2 краплі розчину нітрату кобальту (ІІ), підсушують і спалюють папір. Зола – темно-зелене забарвлення („рінманова зелень”): 2ZnSO4 + 2Co(NO3)2 → 2CoZnO2 + 2SO3↑ +4NO2↑ + O2↑ „рінманова зелень” Умови:
|
Сr3+ |
CrCl3 + 9NH4OH → [Cr(NH3)6](OH)3 + 3NH4Cl + 6H2O Cr3+ + 9NH4OH → [Cr(NH3)6](OH)3 + 3NH4+ + 6H2O |
2CrCl3 + 3H2O2 + 10KOH ® 2K2CrO4 + 6KCl + 8H2O
C відновник 6 2O-1 + 2ē ® 2 O-2 3 відновлення
о 2Cr3+ + 6O-1 ® 2Cr+6 + 6O-2 Умови: лужне середовище, кип’ятіння розчину до повного видалення бульбашок кисню |
|
Мn2+ |
2MnSO4 + 5NaBiO3 + 16HNO3 ® 2HМnO4 + 5Bi(NO3)3+ 2Na2SO4 + NaNO3 + + 7H2O M n2+ - 5ē ® Mn+7 2 окислення відновник 10 Bi+5 + 2ē ® Bi+3 5 відновлення окислювач
2 Умови: азотнокисле середовище, на холоді |
 r+3
- 3ē ®
Cr+6
2
окислення
r+3
- 3ē ®
Cr+6
2
окислення кислювач
кислювач Mn+2
+ 5Bi+5
®
2Mn+7
+
5Bi+3
Mn+2
+ 5Bi+5
®
2Mn+7
+
5Bi+3
Аналіз суміші катіонів ііі аналітичної групи
Попередні дослідження: |
Якщо досліджуваний розчин жовто-коричневого кольору, то присутні іони Fe3+, якщо зеленого – іони Cr3+ |

Відкриття катіона Fe3+: |
Досліджуваний розчин + NH4SCN криваво-червоне забарвлення розчину. Досліджуваний розчин + K4[Fe(CN)6] темно-синій осад |

Відокремлення амфотерних гідроксидів від неамфотерних. Відкриття Cr3+: |
Досліджуваний розчин + NaOH в надлишку осад + Н2О2 (на водяній бані до повного виділення газу) центрифугуванням відокремлюють осад При наявності Сr3+ - центрифугат має жовтий колір |
Виявлення катіона Al3+ в центрифугаті: |
Центрифугат + NH4Cl(кристали) білий осад (після охолодження) |
Виявлення катіона Zn2+: |
Ц ентрифугат + СН3СООН + Н2S білий осад |
Виявлення катіона Mn2+ в осаді: |
Осад промивають водою, відокремлюють центрифугуванням Осад + NaBiO3 (кристали) + HNO3 (концентр.) рожево – фіолетовий розчин |
Лекція №11. Катіони іv аналітичної групи. Аналіз суміші катіонів
Основні питання:
Загальна характеристика катіонів ІV аналітичної групи.
Характеристика групового реактиву.
Загальні реакції катіонів ІV аналітичної групи.
Характерні реакції катіонів ІV аналітичної групи.
Аналіз суміші катіонів ІV аналітичної групи.
Загальна характеристика катіонів іv аналітичної групи
До ІV аналітичної групи належать катіони Ag+, Pb2+, Cu2+. Катіони Ag+, Pb2+ ‑ безбарвні, катіони Cu2+ мають блакитний колір. Ступінь окислення „+1” виявляє аргентум, ступінь окислення ”+2” – у купруму та плюмбуму.
Для катіонів Ag+, Pb2+, Cu2+ характерна здатність до комплексоутворення, координаційні числа – „2” (аргентум), „4” (плюмбум), „6” (купрум).
Основи катіонів ІV аналітичної групи нерозчинні у воді: Сu(ОН)2 – нерозчинна основа, АgOH розкладається водою, Рb(ОН)2 виявляє амфотерні властивості. Нітрати розчинні у воді. Сульфати срібла, свинцю малорозчинні, сульфат міді добре розчиняється у воді. Сульфіди – нерозчинні у воді, розведених кислотах (крім HNO3).
ІV група поділяється на дві підгрупи:
підгрупа срібла (Ag+, Pb2+) – хлориди нерозчинні у воді;
підгрупа міді (Cu2+) – хлорид розчинний у воді.
Груповий реактив – сірководень (H2S) у присутності НСІ (для запобігання осадженню катіонів ІІІ аналітичної групи). Осаджуються сульфіди катіонів ІV аналітичної групи (чорні осади).
Для підкислення розчину використовують НСІ (HNO3 окислює сірководень до вільної сірки):
2HNO3 + 3H2S 3S↓ + 2NO↑ + 4H2O
H2SO4 осаджує іони Pb2+, Ba2+:
H2SO4 + Pb(NO3)2 PbSO4↓ + 2HNO3
Дія H2S сірководню:
2AgNO3 + H2S Ag2S↓ + 2HNO3
2Ag+ + 2NO3- + H2S Ag2S↓ + 2H+ + 2NO3-
2Ag+ + H2S Ag2S↓ + 2H+
Pb(NO3)2 + H2S PbS↓ + 2HNO3
Pb2+ + 2NO3- + H2S PbS↓ + 2H+ + 2NO3-
Pb2+ + H2S PbS↓ + 2H+
Аналогічно відбувається осадження Cu2+.
Розчинення сульфідів в азотній кислоті при нагріванні (помутніння розчину) – утворення вільної сірки:
3Ag2S↓ + 8HNO3 6AgNO3 + 3S↓ + 2NO↑ + 4H2O
3CuS↓ + 8HNO3 3Cu(NO3)2 + 3S↓ + 2NO↑ + 4H2O
Аналогічно відбувається розчинення PbS.
Дія HCl – утворення білих осадів хлоридів срібла та свинцю:
AgNO3 + HСl AgCl↓ + HNO3
Ag+ + Cl- AgCl↓
Аналогічно відбувається осадження Pb2+.
Розчинення AgCl в розчині аміаку – утворення безбарвного розчину діамінаргентум(І) хлорид:
AgCl↓ + NH3∙H2O Ag(NH3)2Cl + 2H2O
Дія лугу в еквівалентній кількості:
2AgNO3 + 2NaOH Ag2O↓ + H2O + 2NaNO3
2Ag+ + 2NO3- + 2Na+ + 2OH- Ag2O↓ + H2O + 2Na+ + 2NO3-
2Ag+ + 2OH- Ag2O↓ + H2O
Pb(NO3)2 + 2NaOH Pb(OH)2↓ + 2NaNO3
Pb2+ + 2OH- Pb(OH)2↓
Аналогічно відбувається осадження Cu2+.
Розчинення Pb(OH)2 у надлишку лугу (амфотерні властивості):
Pb(OH)2↓ + 2NaOH Na2PbO2 + 2H2O
Pb(OH)2↓ + 2Na+ + 2OH- 2Na+ + PbO22- + 2H2O
Pb(OH)2↓ + 2OH- PbO22- + 2H2O
Дія KI – утворення осадів йодидів плюмбуму(ІІ) та купруму (І):
Pb(NO3)2 + 2KI KNO3 + PbI2↓
Pb2+ + 2I- PbI2↓
Аналогічно відбувається взаємодія з катіонами Ag+.
2CuCl2 +4KI 2CuI↓ + I2 + 4KCl
2Cu2+ + 4I- 2CuI↓ + I2↓
Дія надлишку NH4OH – осад основної солі свинцю, утворення комплексних аміакатів Ag+,Cu2+:
Ag2O↓ + 4NH4OH 2Ag(NH3)2OH + 3H2O
Pb(NO3)2 + NH4OH PbOHNO3↓ + NH4NO3
Pb2+ + NH4OH PbOH+ + NH4+
2CuSO4 + 2NH4OH Cu(OH)2↓ + (NH4)2SO4
2Cu2+ + SO42- + 2NH4OH Cu(OH)2↓ + 2(NH4)+
Cu2(OH)2SO4↓ + 10NH4OH + (NH4)2SO4 12H2O + 2Cu(NH3)6SO4
Cu2(OH)2SO4↓ + 10NH4OH + 2NH4+ 12H2O + 2Cu(NH3)62+
Дія хромату калію:
K2CrO4 + 2AgNO3 Ag2CrO4↓ + 2KNO3
CrO42- + 2Ag+ Ag2CrO4↓
Аналогічно відбувається осадження Pb2+.
Загальні реакції катіонів ІV аналітичної групи
Реактиви |
Катіони |
||
Ag+ |
Pb2+ |
Cu2+ |
|
H2S сірководень |
Ag2S чорний осад |
PbS чорний осад |
CuS чорний осад |
Всі осади розчинні в HNO3 при нагріванні. |
|||
HCl |
AgCl білий осад розчинний в розчині аміаку Ag(NH3)2Cl безбарвний розчин |
PbCl2 білий осад, розчинний в гарячій воді |
- |
Осади нерозчинні в HNO3 |
|||
NaOH в еквівалентній кількості |
Ag2O чорно-коричневий осад |
Pb(OH)2 білий осад |
Cu(OH)2 блакитний осад |
NaOH в надлишку |
Ag2O чорно-коричневий осад |
K2[Pb(OH)4] безбарвний розчин |
Cu(OH)2 блакитний осад |
NH4OH в еквівалентній кількості та в надлишку |
Ag2O чорно-коричневий, поступове розчинення Ag(NH3)2OH безбарвний розчин |
Pb(OH)NO3 білий осад |
Cu2(OH)2SO4 зеленувато-блакитний осад, поступове розчинення Cu(NH3)6SO4 темно-синій розчин |
KI |
AgI жовтий осад |
PbI2 жовтий кристалічний осад, розчиняється в гарячій воді |
CuI2 розкладається CuI білий осад I2 – бурий |
K2CrO4 |
Ag2CrO4 цегляно-червоний осад |
PbCrO4 жовтий осад |
- |
Осади добре розчинні в HNO3 |
|||
|
розчиняється в аміаку |
розчин. в надлишку лугу |
|
Характерні реакції катіонів ІV аналітичної групи
Катіон |
Характерна реакція та умови її проведення |
Аg+ |
AgNO3 + HCl ® AgCl↓ + HNO3 Ag+ + Cl- ® AgCl↓ Осад розчиняється в концентрованій HCl, водному розчині аміаку з утворенням комплексних сполук: AgCl↓ + NH3∙H2O Ag(NH3)2Cl + 2H2O |
AgNO3 + KI ® AgI↓ + KNO3 Ag+ + I- ® AgI↓ Осад не розчиняється в NH3∙H2O, HNO3, проте розчиняється у надлишку КІ з утворенням К[AgI2]: AgI↓ + КІ → К[AgI2] AgI↓ + І- → [AgI2]- |
|
Рb2+ |
Pb(NO3)2 + 2KI ® 2KNO3 + PbI2↓ Pb2+ + 2I- ® PbI2↓ Розчинення PbI2 в надлишку КІ: PbI2↓ + 2KI ® K2[PbI4] PbI2↓ + 2I- ® [PbI4]2- |
Сu2+ |
CuSO4 + 4NH4OH ® [Cu(NH3)4(H2O)2]SO4 + 2H2O Cu2+ + 4NH4OH ® [Cu(NH3)4(H2O)2]2+ + 2H2O |
Катіони Cu2+ забарвлюють полум’я пальника в зелений колір. |
Біохімічна роль катіонів іv аналітичної групи
Мідь (купрум) міститься в організмі у сполуках з білками в еритроцитах (гемокупрен), у плазмі крові (церулоплазмін), металотіонеїн (білок, що відповідає за відкладання міді).
Добова потреба – 2мг. Джерела міді : печінка, яєчні жовтки, зелені овочі.
Роль міді:
участь в утворенні еритроцитів;
вивільненні тканинного заліза і розвитку скелету, центральної нервової системи.
Надлишок міді викликає подразнення та роз’їдання слизових оболонок; порушення у роботі печінки, нирок, капілярів, центральної нервової системи.
Нестача міді: слабкість артерій, порушення діяльності печінки, вторинна анемія.
Ряд металів, таких як, залізо, мідь, навіть в невеликих концентраціях можуть викликати небажане окислення продуктів. Їх каталітична окислювальна здатність особливо яскраво виявляється відносно жирів і жирових продуктів. При концентрації заліза вище 1,5мг/кг та міді 0,4мг/кг при тривалому зберіганні вершкового масла і маргаринів ці метали викликають згіркнення продуктів. При зберіганні напоїв у присутності заліза вище 5 мг/л та міді 1мг/л за певних умов може спостерігатися помутніння напоїв.
Свинець (плюмбум) – один з найпоширеніших і небезпечних токсикантів. Поширеність у земній корі – 1,6·10-3%. Сполуки свинцю – Pb3O4 PbSO4 – основа пігментів сурику та свинцевих білил. Світове добування свинцю складає понад 3,5·106т на рік. У природні води кожний рік потрапляє 500-600 тис. т, а в атмосферу в переробленому та дрібнодисперсному стані викидається близько 450 тис.т свинцю. Більша кількість свинцю осідає на поверхні землі.
Добове надходження свинцю в організм людини з їжею складає 0,1 – 0,5 мг; з водою – 0,02 мг. ГДД свинцю складає 0,007 мг/кг; величина в питній воді – 0,05 мг/л.
Вміст свинцю (мг/кг) в різних продуктах такий: фрукти 0,01 – 0,6; овочі 0,02 – 1,6; крупи 0,03 – 3,0; хлібобулочні вироби 0,03 – 0,83; м’ясо та риба 0,01 - 0,78; молоко 0,01 – 0,1.
В організмі людини засвоюється в середньому 10% свинцю, у дітей – 30 – 40%. З крові свинець надходить до м’яких тканин і кісток, де депонується у вигляді трифосфату.
Механізм токсичної дії свинцю:
Блокування функціональних SH-груп білків. як результат – інактивація ферментів.
Проникнення свинцю у нервові та м’язові клітини, утворення лактату свинцю, потім фосфату свинцю, які створюють клітинний бар’єр для проникнення іонів кальцію.
Свинець діє на кровотворну, нервову та перетравлювальну системи організму, нирки.
Свинцева інтоксикація викликає порушення здоров’я:
головні болі головокружіння;
підвищена втомлюваність, роздратованість, погіршення сну;
м’язова гіпотонія; параліч і парез;
розумова відсталість.
Дефіцит кальцію, фосфору, заліза, пектинів, білків, підвищене надходження кальциферолу підвищують засвоюваність свинцю (його токсичність).
Аналіз суміші катіонів ІV аналітичної групи
Виявлення катіону Cu2+: |
Досліджуваний розчин (блакитний колір) + NH4OH (надлишок) ® синьо - фіолетовий розчин |
Виявлення підгрупи срібла: |
Досліджуваний розчин + HCl ® білий осад |
Осадження катіонів підгрупи срібла: |
Досліджуваний розчин + HCl ® білий осад (І), який відокремлюють центрифугуванням ® перевірка на повноту осадження (HCl) ® відсутність осаду. При наявності Cu2+ - осад відокремлюють, промивають холодною водою, промивні води відкидають. |
Виявлення Pb2+: |
Осад (І) + H2O (дистильована) – нагрівання на водяній бані → додають КІ ® жовті кристали PbI2 |
Відокремлення Pb2+: |
Осад (І) + H2O (дистильована) – нагрівання на водяній бані декілька разів → центрифугування ® перевірка на повноту осадження (КІ). |
Виявлення катіону Ag+: |
Осад (ІІ) + NH4OH ® розчинення осаду (ІІ) ® розчин (ІІ) + КІ ® жовтий осад AgI розчин (ІІ) + HNO3 ® білий осад AgCl |
Лекція №12. Аніони і-ііі аналітичних груп
Основні питання:
Класифікація аніонів.
Якісні реакції аніонів І-ІІІ аналітичних груп.
Аналіз суміші аніонів.
Класифікація аніонів
Аналітична група |
Груповий реактив |
Характеристика групи |
Приклади аніонів |
І |
BaCl2 в нейтральному або слабколужному середовищі |
Осади, що розчиняються в HNO3 (виняток BaSO4). З розчином AgNO3 – осади, що розчиняються в HNO3 |
SO42-, CO32-, SO32-, SiO32-, PO43- |
ІІ |
AgNO3 |
Осади, що не розчинні в HNO3 З розчином BaCl2 – не утворюють осаду |
Cl-, Br-, I-, S2- |
ІІІ |
немає |
З розчинами AgNO3 та BaCl2 ‑ осади не утворюються |
NO3-, NO2- |
Нітрати натрію використовуються як харчові добавки (консерванти) до певних сортів сиру (0,01-0,02% додають до молока для уповільнення спучування), а також до м’ясопродуктів та рибопродуктів. Нітрати можуть перетворюватися на нітрити у ході ферментативних процесів і під впливом мікроорганізмів. Відновлення відбувається самочинно в продуктах харчування та перетравлю вальній системі людини. У дорослих перетворення нітратів до нітритів відбувається в кишках, у немовлят – у шлунку або дванадцятипалій кишці (легке всмоктування нітратів). Летальна доза нітрату складає30-35 мг на 1 кг маси тіла; ГДД 0 – 5 мг на 1 кг маси тіла (для дітей – нижча).
Летальна доза нітриту складає 32мг на 1кг маси тіла, тобто близько 2г. Нітрити виявляють мутагенну дію. Нітрити не тератогенні, однак не виключена можливість утворення канцерогенних N-нітрозосполук з нітритів і амінів.
Нітрити натрію та калію (Е249, Е250) використовують в основному в суміші з повареною сіллю для засолу м’ясопродуктів і як консерванти для них. Антимікробна дія виявляється при концентраціях 50-160 мг на 1 кг продукту. Додавання нітритів до ковбас і копченостей сприяє утворенню необхідного кольору і специфічного аромату, уповільнює ріст патогенних і токсичних мікроорганізмів. Нітрити легко всмоктуються з кишково-шлункового тракту, знижують тонус гладкої мускулатури, розширюють судини і знижують кров’яний тиск. У більш високих концентраціях нітрити утворюють метгемоглобін і викликають ціаноз.
Реакції аніонів І аналітичної групи
Аніон |
Характерна реакція та умови її проведення |
SO42- (сульфат-аніон) |
BaCl2 + Na2SO4 ® BaSO4↓ + 2NaCl Ba2+ + SO42- ® BaSO4↓ |
SO32- (сульфіт-аніон) |
BaCl2 + Na2SO3 ® BaSO3↓ + 2NaCl Ba2+ + SO32- ® BaSO3↓ Осад розчиняється в мінеральних кислотах, крім сульфатної: BaSO3↓ + 2HNO3 ® Ba(NO3)2 + SO2↑ + H2O BaSO3↓ + 2H+ ® Ba2+ + SO2↑ + H2O |
Na2SO3 + 2HCl ® SO2↑ + H2O + 2NaCl SO32- + 2H+ ® SO2↑ + H2O SO2 виявляють за знебарвленням розчинів І2, KMnO4 (SO2 – відновник): SO2 + I2 + 2H2O ® H2SO4 + 2HI 5SO2 +2KMnO4 + 2H2O ® K2SO4 + 2MnSO4 + 2H2SO4 |
|
Na2S+4O3 + I20 + H2O ® Na2S+6O4 + 2HI- S+4 -2e → S+6 I20 +2e → 2І- 5Na2S+4O3 + 2KMn+7O4 + 3H2SO4 ® 5Na2S+6O4 + 2Mn+2SO4 + K2SO4 + 3H2O S Mn+7 +5е → Mn+2 2 5S+4 + 2Mn+7 → 5S+6 + 2Mn+2 |
|
Аніон |
Характерна реакція та умови її проведення |
CO32- (карбонат-аніон) |
BaCl2 + Na2CO3 ® BaCO3↓ + 2NaCl Ba2+ + CO32- ® BaCO3↓ BaCO3↓ + 2HCl ® BaCl2 + CO2↑ +H2O BaCO3↓ + 2H+ ® Ba2+ + CO2↑ +H2O |
Na2CO3 + 2HCl ® CO2↑ +2NaCl + H2O CO32- + 2H+ ® CO2↑ + H2O При пропусканні CO2 крізь розчин гідроксиду кальцію – утворення білого осаду: CO2↑ + Ca(OH)2 ® CaCO3↓ + H2O |
 +4
-2e
→ S+6
5
+4
-2e
→ S+6
5Реакції аніонів ІI аналітичної групи
Аніон |
Характерна реакція та умови її проведення |
Cl- (хлорид-аніон) |
AgNO3 + NaCl ® AgCl↓ + NaNO3 AgCІ – не розчиняється в розведених кислотах, але розчиняється в розчині аміаку: AgCl↓ + 2NH4OH ® [Ag(NH3)2]Cl + 2H2O |
S2- (сульфід-аніон) |
2AgNO3 + Na2S ® Ag2S↓ + 2NaNO3 2Ag+ + S2- ® Ag2S↓ Ag2S не розчиняється в розчині аміаку, але розчиняється при нагріванні в HNO3: 3Ag2S-2↓ + 8HN+5O3 ® 6AgNO3 + 3S0↓ + 2N+2O↑ + 4H2O S N+5 +3e→ N+2 2 3S-2 + 2N+5 → 3S0 + 2N+2 |
Аніон |
Характерна реакція та умови її проведення |
S2- (сульфід-аніон) |
Na2S + 2HCl ® 2NaCl +H2S↑ S2- + 2H+ ® H2S↑ H2S виявляють за почорнінням фільтрувального паперу, змоченого розчином ацетату свинцю: H2S↑ + Pb(CHCOO)2 ® PbS↓ + 2CH3COOH H2S↑ + Pb 2+ ® PbS↓ +2Н+ |
 -2
‑2e
→
S0
3
-2
‑2e
→
S0
3Реакції аніонів ІII аналітичної групи
Аніон |
Характерна реакція та умови її проведення |
NO3- (нітрат-аніон) |
2NaN+5O3 + 6Fe+2SO4 + 4H2SO4 ® 3Fe2+3(SO4)3 + Na2SO4 + 2N+2O↑ + 4H2O N +5 +3е → N+2 2 2Fe+2 -2е → 2Fe+3 3 2N+5 + 6Fe+2 → 2N+2 + 6Fe+3 2NO + O2 ® 2NO2 |
NO2- (нітрит-аніон) |
2KMn+7O4 + 5NaN+3O2 + 3H2SO4 ® 5NaN+5O3 + 2Mn+2SO4 + K2SO4 + 3H2O M n+7 +5е → Mn+2 2 N+3 -2е → N+5 5 2Mn+7 + 5N+3 → 2Mn+2 + 5N+5 NaNO2 ‑ відновник |
Аналіз суміші аніонів І-ІІІ аналітичних груп
Попередні дослідження: |
Випробування реакції досліджуваного розчину:
|
Виявлення аніонів І аналітичної групи: |
Досліджуваний розчин + BaCl2 ® білий осад ® центрифугування; Осад + HNO3 ® розчинення осаду (відсутність SO42-) |
|
|
|
|
Виявлення аніонів ІІ аналітичної групи: |
Досліджуваний розчин + HNO3 + AgNO3 ® білий осад |
|
|
|
|
Виявлення аніонів ІІІ аналітичної групи: |
|
|
|
|
Після припинення виділення газу + дифеніламін ® синє забарвлення. |
Лекція №13. Кількісний аналіз. Методи кількісного аналізу
Основні питання:
Кількісний аналіз.
Методи кількісного аналізу.
Класифікація хімічних методів кількісного аналізу.
Кількісний аналіз – визначення кількісного вмісту елементів, іонів або хімічних сполук, що входять до складу досліджуваних речовин і матеріалів.
Предметом кількісного аналізу є вивчення методів визначення кількісного складу речовини і науково-теоретичне обґрунтування цих методів.
Завданням кількісного аналізу є визначення масового (вагового) співвідношення елементів, які входять до складу досліджуваної системи.

Умови проведення кількісного аналізу:
Правильний вибір аналітичної реакції або фізичних властивостей речовин;
Правильне виконання аналітичних операцій;
Використання надійних способів вимірювання результатів аналізу.
К ількісний
аналіз
– це основний засіб визначення якості
речовин і матеріалів, що залежить від
кількості основних компонентів, домішок.
ількісний
аналіз
– це основний засіб визначення якості
речовин і матеріалів, що залежить від
кількості основних компонентів, домішок.
Види кількісного аналізу:
аналіз неорганічних речовин;
аналіз органічних речовин.
За допомогою кількісного аналізу можна визначити відповідність характеристик речовин або матеріалів вимогам нормативно-технічних документів, які розробляють на основі результатів аналізу стандартних зразків (СЗ) речовин і матеріалів, які відповідають певним потребам споживача.
Стандартні зразки – різні матеріали, вміст окремих елементів у яких відомий з великою точністю. Використовують у різних аналітичних методах: для побудови калібрувальних графіків і встановлення таким чином складу речовин і контролю правильності аналізу, для об’єктивної метрологічної характеристики різних методів аналізу. При ідентифікації досліджуваного зразка зі стандартним зразком визначають кількість складових частин досліджуваного зразка і його фізичні константи.

Методи кількісного аналізу використовують для:
перевірки правильності проведення технологічних процесів (поетапний контроль);
розв’язання питань дослідницько-прикладного характеру (присутність токсичних речовин у продуктах харчування, навколишньому середовищі);
встановлення складу і будови речовин;
встановлення швидкості та механізмів перебігу реакцій.
Роль хімічного аналізу:
З’ясування природи досліджуваної речовини;
Визначення складу та вмісту основного компоненту та сторонніх домішок;
Встановлення хімічної формули невідомої речовини;
Встановлення структури речовини.
За кількістю речовини, що взята для аналізу, аналітичні методи поділяють:
Макроаналіз (1 ‑ 10г твердої речовини, 10 ‑ 100мл розчину);
Напівмікроаналіз (0,05 ‑ 0,5г твердої речовини, 1 ‑ 10мл розчину);
Мікроаналіз (0,001 ‑ 1 10-6г твердої речовини, 0,1 – 10-4мл розчину).
Проба – це частина досліджуваного матеріалу, що відображає його хімічний склад. У пробі розрізняють визначувану речовину або кількість визначуваних речовин і основу.
Представницька проба – це відповідність складу проби середньому матеріалу, що аналізують. Результат аналізу може бути правильним лише тоді, коли проба досить представницька (точно відображає склад матеріалу, з якого вона відібрана).
Методи відбору представницької проби регламентовані державними стандартами і залежать від характеру матеріалу: рідини перемішують, використовують спеціальні пробовідбірники, які занурюють на певну глибину і захоплюють ними порції рідини; проби в’язких матеріалів відбирають після старанного перемішування з верхньої, середньої і нижньої частин маси; проби твердих і сипких матеріалів відбирають з різних місць упаковки так, щоб були захоплені зовнішні і внутрішні шари матеріалу; при відборі проби з кількох ємностей рідину в кожній з них перемішують, відбирають з кожної посудини однакові об’єми рідин і змішують їх; рідини з осадом ретельно перемішують, що осад рівномірно розподілився по всьому об’ємі рідини, і швидко відбирають пробу.
Відібрана середня проба речовини зберігається в герметично закритій тарі, і береться в міру потреби для проведення аналізу.
Види проб:
первинна (генеральна) – відбирається на першому етапі від великої маси матеріалу і зменшується до розмірів лабораторної проби квартуванням;
лабораторна (паспортна) – одержують при зменшенні генеральної проби до маси, необхідної для проведення повністю всього аналізу. Маса лабораторної проби залежить від вмісту визначуваної речовини і чутливості методики аналізу (чим чутливіша методика аналізу, тим менша маса лабораторної проби);
аналітична проба – відбирають від лабораторної проби для проведення аналітичного визначення.
На точність кількісного аналізу впливає:
маса речовини або наважка, що взята для аналізу;
об’єм розчину, що вимірюють мірним посудом.
Абсолютно точні результати аналізу практично неможливо одержати, за реальних умов визначають відтворюваність аналізу, що залежить від кількості вимірювань. Відтворюваність обчислюється на основі паралельних аналізів і характеризує випадкові погрішності. Внаслідок неповноти проходження хімічної реакції виникають хімічні погрішності.
Кінцевий результат кількісного визначення виражається числом, що вказує вміст визначуваного компонента в досліджуваному зразку. Цей результат одержують після декількох послідовно проведених хімічних операцій (розчинення, осадження, фільтрування, висушування, прожарювання тощо), що сильно впливає на точність отриманого результату.
Помилку може спричиняти неправильне відбирання середньої проби досліджуваної речовини. В аналізі одержують величини, які вимірюються з певними помилками. Їх називають погрішностями визначення. Якщо погрішності визначення однакові за величиною але протилежні за знаком, то помилка виключається.
Помилки поділяють на:
систематичні помилки залежать від постійно діючих причин та повторюються при всіх відліках. До них належать помилки інструментів (терези, бюретки, піпетки, індивідуальні помилки спостерігача, помилки вибраного методу).
випадкові помилки визначають випадковими причинами, спричиняються недосконалістю приладів і органів чуття спостерігача. Теорія помилок дозволяє зменшити вплив випадкових помилок на кінцевий результат вимірювання та достатньо точно встановити можливу помилку.
грубі помилки пов’язані з невірним відліком та недостатньо ретельним проведенням аналітичних операцій. Величини, одержані з грубими помилками відкидаються.
Кожному методу аналізу притаманні свої помилки, які можуть бути відсутніми в інших методах. Наприклад, помилки, пов’язані з втратою речовини під час прожарювання, спостерігаються в гравіметричному аналізі, проте відсутні в титриметричному. Помилки, пов’язані із використанням індикаторів, характерні для титриметричного аналізу, проте відсутні в гравіметричному. Є помилки, які характерні для всіх методів кількісного аналізу. Наприклад, при зважуванні на аналітичних терезах, можна завжди допустити помилку, що дорівнює ±0,0002г. В ретельно проведеному аналізу неорганічних речовин відносна помилка не повинна перевищувати ± 0,1%. Тому наважка речовини для аналізу не повинна бути меншою ніж 0,2г.
В результатах аналітичного визначення завжди є погрішність. Оцінка погрішності є частиною аналізу, а сама погрішність – важлива характеристика аналізу.
Погрішності в кількісному аналізі:
Випадкові (недоброякісне виконання операцій), можуть мати різні числові значення, передбачити їх неможливо, можуть бути зменшені збільшенням числа паралельних визначень. Випадкові погрішності не можуть бути усунені, але їх вплив може бути врахований статистичною оцінкою результатів аналізу, характеризують відтворюваність методики.
Систематичні (неправильний вибір методу аналізу, недосконалість приладів, неправильні розрахунки, записи) – ті, що повторюються при повторних визначеннях погрішностей. Числові значення їх однакові при всіх визначеннях, які проводились одним і тим же методом (часто розглядають як показник високої точності і правильності аналізу). Для з’ясування систематичної погрішності проводять аналіз цього ж зразка іншими методами. Систематична погрішність характеризує правильність аналізу.
Погрішність вимірювання (абсолютні та відносні помилки) – це відхилення результату вимірювання від справжнього значення вимірюваної величини.
Абсолютна похибка (Δ) – різниця між експериментально отриманим значенням (Хексп.) та істинним вмістом компоненту (Х іст.). Залежить від точності калібрування мірного посуду, обумовлена погрішністю аналітичних терезів, класом точності приладів, методами вимірювання, індивідуальними особливостями спостерігача.
Δ
=
![]()
![]()
Відносна
похибка (![]() )
– відношення абсолютної помилки (Δ) до
істинного вмісту компоненту (Хіст.)
)
– відношення абсолютної помилки (Δ) до
істинного вмісту компоненту (Хіст.)
=
![]()
Класифікація хімічних методів кількісного аналізу

Лекція №14. Гравіметричний (ваговий) метод аналізу
Основні питання:
Сутність гравіметричного аналізу.
Методи гравіметричного (вагового аналізу).
Основні операції гравіметричного аналізу та послідовність їх виконання.
Гравіметричний аналіз базується на законах збереження маси та сталості складу речовини, полягає в точному вимірюванні маси досліджуваного компоненту, який був отриманий у вигляді сполуки певного хімічного складу.
Гравіметричний аналіз ґрунтується на хімічних законах:
збереження маси;
сталості складу речовини;
еквівалентів.
Мета гравіметричного аналізу – визначення масової частки (%) елементної речовини в будь-якій речовині або в суміші речовин чи складних систем.




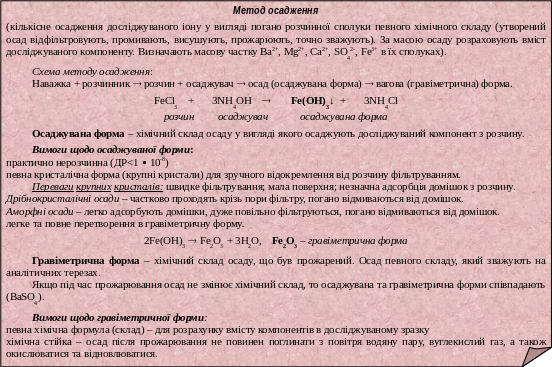
Операції вагового (гравіметричного) аналізу
(незначна
кількість досліджуваного матеріалу,
що за своїми фізичними та хімічними
властивостями, складом, відповідає
всій партії). Розрізняють
3 види середніх проб: первинна
(відбирається
з партії матеріалу); лабораторна
або паспортна (отримують зменшенням
первинної проби до маси, необхідної
для здійснення всіх аналізів),
подрібнюють; аналітична
(відбирають з лабораторної для проведення
хімічного аналізу). Квартування.
Зберігають в бюксах з кришкою.І. Відбір середньої проби
Маса
наважки залежить від властивостей
речовини і методики аналізу (якщо
наважка дуже мала, збільшиться погрішність
аналізу, якщо дуже велика – аналіз
займе багато часу).
Наважка(0,1
–1,0г) – відбирається з аналітичної
середньої проби досліджуваного зразка. Зважується
приблизно на технохімічних терезах, а
потім з точністю до 0,0001г на аналітичних.
Зважують наважку на часових скельцях
або в бюксах з кришкою (гігроскопічні
речовини).
Способи
взяття наважки: Спочатку
зважують порожній бюкс, беруть необхідну
масу речовин, зважують бюкс з речовиною.
Речовину обережно кількісно переносять
у стакан, в якому будуть розчиняти
наважку, змиваючи туди ж залишки
речовини з бюкса струменем води з
промивалки. Різниця між масою бюкса з
речовиною і масою порожнього бюкса є
величина взятої наважки. Зважують
порожній бюкс. Потім зважують бюкс з
речовиною. Після цього речовину обережно
переносять у стакан або колбу і бюкс
із залишками речовини, знову зважують.
Різниця між масою бюкса з речовиною і
масою бюкса з залишками речовини дає
наважку. Наважку
переносять в стакан, додають розчинник,
нагрівають на водяній бані, накривши
фарфоровою чашкою.Іі. Зважування та розчинення наважки
Проводять
в хімічних стаканах. Умови
осадження (для отримання крупних
кристалів): з
гарячих розведених розчинів гарячим
розведеним розчином осаджувача; осаджувач
додають повільно, по краплям, щоб
запобігти розбризкування; розчин
перемішують скляною паличкою – зменшує
кількість центрів кристалізації,
сприяє росту крупних кристалів; осад
залишають для дозрівання (дрібні
кристали розчиняються, відбувається
ріст кристалів, часткове звільнення
осаду від захоплених ним з розчину
сторонніх іонів)Ііі. Осадження
Проводять: після
осадження (після повного охолодження
розчину), якщо осад погано розчинний
– кристалічні осади. Аморфні осади
одразу фільтрують; через
деякий час, якщо розчинність осаду
збільшується з підвищенням температури.
Для
відокремлення осадів використовують
беззольні фільтри (вміст золи після
спалювання 0,0001г). Беззольні
фільтри: Найбільш
щільні (рожева, чорна та червона стрічки)
- для відокремлення аморфних осадів. Найменш
щільні (біла стрічка) – для відокремлення
крупнокристалічних осадів. Середньої
щільності (блакитна, синя, зелена
стрічка) – для відокремлення
дрібнокристалічних осадів.
Розмір
фільтра залежить від осаду (1/3 фільтра),
край фільтра повинен бути на 5-7 мм нижче
краю лійки. Лійка (скляна) – на 0,5-1см
вище краю фільтра. Правильне
фільтрування: Фільтр
складають, вставляють в лійку, змочують
водою; Рівень
рідини в фільтрі – на 1-2см не доходить
до краю паперу; Переливають
рідину на фільтр по скляній паличці,
не можна змочувати осад на дні стакану; Додаючи
воду до осаду, його взмучують, дають
відстоятися (декантація). Переносять
осад на фільтр. Проба
на повноту відмивання (промивні води
не
дають реакцію на досліджувані іони).Іv. Фільтрування (відокремлення осаду від маточного розчину)
Відмитий
від домішок осад в лійці підсушують в
сушильній шафі (t=90-1050С),
накривши лійку з осадом листком паперу,
який в кількох місцях проколотий гострим
кінцем скляної палички.
Висушений
фільтр з осадом дістають з лійки,
згортають пакетиком, вміщують в тигель
конусом вниз, тигель з осадом поміщають
у фарфоровий трикутник у похилому
положенні над пальником, обвуглюють
фільтр на газовому пальнику, причому
полум’я пальника роблять таким, щоб
при легкому нагріванні краєм полум’я
папір тлів, а не згорав (проводять
озоління), тигель переносять тигельними
щипцями в муфельну піч та прожарюють
протягом 1 год. при t=600-8000С.
Мета
прожарювання:
видалення розчинника і летких
електролітів, розклад осаду з утворенням
продуктів відомого складу. Потім тигель
охолоджують в ексикаторі.
Ексикатор
– це спеціальний посуд, який використовують
для захисту предметів від поглинання
вологи з повітря. У нижній частині
ексикатора знаходиться осушуюча
речовина.V. Висушування та прожарювання
Тигель
зважують на аналітичних терезах. Прожарювання
повторюють (результати двох зважувань
не повинні відрізнятися більше 0,0002г).
Після
доведення осаду до постійної маси
приступають до обчислень результатів
аналізу.
VI. Зважування
VII. Обчислення результатів аналізу Лекція № 15. Титриметричний (об’ємний) метод аналізу. Метод нейтралізації
Основні питання:
Сутність титриметричного (об’ємного) аналізу. Титрування, виготовлення розчинів в титриметричному аналізі.
Вимірювальний посуд та його призначення в об’ємному аналізі.
Операції титриметричного визначення.
Методи об’ємного аналізу.
Сутність методу нейтралізації. Індикатори: інтервал переходу, показник титрування, вибір індикатору. Застосування методу.
Об’ємний аналіз – метод кількісного аналізу, що базується на точному вимірюванні кількості (об’єму) розчину реагенту точно відомої концентрації, витраченому на реакцію з еквівалентною кількістю досліджуваної речовини.
Реагент – розчин з точно встановленою концентрацією (стандартний розчин, робочий титрований розчин або робочий розчин). Концентрацію виражають через його молярну концентрацію речовини еквівалента (нормальність), молярність, титр.
Процес додавання робочого розчину реагенту до досліджуваного розчину називається титруванням.
Розчин, що містить активний реагент, за допомогою якого проводять титрування, називається титрант.
Титрант поміщають у бюретку, а розчин, який титрують – титрований розчин – у конічну колбу.
Титрують до досягнення точки еквівалентності, тобто моменту, коли кількість реактиву, яка є в об’ємі робочого розчину, стане еквівалентною кількості досліджуваної речовини.
Методи визначення точки еквівалентності:
за допомогою спеціальних індикаторів (речовин, здатних змінювати своє забарвлення після введення робочого розчину в еквівалентній кількості);
за забарвленням реагуючих речовин;
за допомогою спеціальних приладів.
Точка кінця титрування - момент титрування, коли індикатор змінює своє забарвлення.
Практично неможливо підібрати індикатор, який показував би кінець титрування в точці еквівалентності. Кінцева точка титрування завжди відрізняється від точки еквівалентності. Чим більша різниця між цими точками, тим більша помилка титрування.
Необхідно підбирати такий індикатор і умови титрування, щоб кінцева точка титрування була якнайближчою до точки еквівалентності.
Умови проведення титриметричного аналізу:
наявність робочого розчину з точно відомою концентрацією;
точне вимірювання об’єму титранту при титруванні бюреткою і об’єму досліджуваного розчину мірною піпеткою;
точне визначення точки еквівалентності.
Вимоги щодо реакції в титриметрії:
реакція повинна бути необоротною – практично відбуватися до кінця;
реакція між досліджуваною речовиною та реагентом повинна відбуватися миттєво (точна фіксація точки еквівалентності), стехіометрично (відповідно до рівняння хімічної реакції);
в досліджуваному розчині не повинні бути наявними інші речовини, які реагують з робочим розчином (відсутність побічних реакцій);
точка еквівалентності повинна легко і точно фіксуватися.
Переваги титриметричного аналізу:
швидкість кількісного визначення речовини;
відносна простота виконання операцій;
можливість використання різних хімічних властивостей речовини (кислотно-основних, окисно-відновних, здатність до осадження і комлексоутворення);
достатня точність.

Стандартизація ‑ це процес знаходження концентрації активного реагенту у розчині, який здійснюється титруванням відомої кількості речовини.
Стандартний розчин – це розчин з точно відомою концентрацією хімічно активної речовини або з точно відомим титром.
Способи виготовлення розчинів в титриметричному аналізі
Безпосереднє зважування потрібної кількості речовини і розчинення взятої наважки в певному (точному) об’ємі розчинника.
Хімічні сполуки, які придатні для виготовлення титрованих розчинів безпосереднім зважуванням препарату, називаються вихідними речовинами (первинні стандартні речовини).
Вимоги щодо вихідних речовин:
чисті або повинні підлягати очищенню (перекристалізацією);
формула повинна точно відповідати певному хімічному складу;
бути стійкою в сухому стані або в розчині;
мати велику молярну масу речовини еквівалента.
Приклади: бура Na2B4O7 ∙ 10H2O; щавлева кислота Н2С2О4 ∙ 2Н2О.
Для приготування первинних стандартних розчинів використовують первинні стандартні речовини і мірні колби.
З фіксаналів (стандарт-титрів) – кількісне розчинення наважки стандарт-титру в мірній колбі місткістю 1л. Таким способом виготовляють 0,1Н розчини (в ампулі міститься 0,1моль речовини еквівалента).
Стандарт-титр – це точна наважка сухої вихідної речовини (або точно відміряна кількість розчину речовини відомої концентрації), яка вміщена в запаяну скляну ампулу, розрахована для приготування розчинів об’ємом 1дм3 (1л) з молярною концентрацією речовини еквівалента 0,1000 моль/л.
Фіксанали можуть бути використані для приготування розчинів вихідних речовин або робочих (NaOH, Na2S2O3).
 Термін
зберігання стандарт – титрів залежить
від природи речовини.
Термін
зберігання стандарт – титрів залежить
від природи речовини.

.
За способом виконання операцій титрування:
Спосіб піпеткування (готують точний об’єм аналізованого розчину, для титрування беруть аліквотний об’єм цього розчину);
Спосіб окремих наважок (беруть всю наважку досліджуваної речовини, яку аналізують).
Вимірювальний посуд в титриметрії
Мірні циліндри – для приблизного відмірювання об’єму розчину.
Мірні колби – це колби з подовженими шийками, відносно малого діаметра, з пришліфованими скляними пробками. На шийці колби позначена її місткість у см3 або в мл і температура (200). Використовують для приготування стандартних і робочих розчинів, розчинів речовин, які аналізують, для точного розведення розчинів в декілька разів. Об’єм мірної колби розрахований на вливання (на шийці одна колова риска), тобто цифра на штампі відповідає об’єму розчину, що міститься в колбі. Такі мірні колби непридатні для відмірювання певних об’ємів розчинів. Колби з двома коловими рисками калібровані одночасно на вливання (нижня риска) і виливання (верхня риска). Перед використанням колба споліскується дистильованою водою. Не можна видувати краплі рідини, яка залишилася в піпетці (градуйовані на виливання).
Піпетки – для точного відмірювання та перенесення в іншу посудину частини досліджуваного розчину. Піпетки калібровані на виливання, тобто цифра на штампі відповідає об’єму розчину, який виливається з піпетки при вільному витіканні рідини. Піпетки бувають циліндричні (градуйовані) – використовують для приблизного відмірювання об’єму розчинів, і з розширенням (прості) – мають одну колову риску. Найбільш поширені піпетки місткістю 1, 2, 5, 10, 25, і 50 мл (см3). Перед використанням піпетки споліскують дистильованою водою, а потім розчином, який відбирають.
Бюретки – це циліндрична трубка з краном або гумовим затвором чи кулькою і нанесеними по всій її довжині поверхні великими і малими поділками. Бюретки призначені для титрування, для точного відмірювання об’єму розчину, витраченого на титрування. Дозволяє вимірювати об’єм розчину з точністю до 0,02-0,03 см3. Об’єм бюреток розрахований на виливання. Нульова поділка розміщена у верхній частині бюретки. Для вимірювання об’ємів від 5 мл до десятих частин мл використовують мікробюретки. Закріплюється на спеціальній стойці або на штативі за допомогою укріплень. Перед використанням бюретка споліскується дистильованою водою, а потім робочим розчином.
Етапи титриметричного (об’ємного) аналізу і. Приготування стандартних і робочих розчинів реагентів
Розрахунок і зважування наважок
Наважку для приготування стандартного розчину вихідної речовини зважують на аналітичних терезах з точністю до 0,0001г, наважку для приготування робочого розчину, точна концентрація якого встановлюватиметься по стандартному розчину, зважують приблизно на технохімічних терезах.
Розчинення наважки
Зважену наважку вихідної речовини переносять в мірну колбу через конічну лійку і доливають воду. В міру розчинення вміст колби переміщують, доливають водою до риски. Виготовлений розчин вихідної речовини використовують для встановлення точної концентрації робочого розчину, який готують приблизної концентрації.
Іі. Встановлення точної концентрації робочого розчину титруванням по вихідній речовині
В бюретку наливають робочий розчин речовини (приблизна концентрація). В конічну колбу для титрування переносять мірною піпеткою аліквотний об’єм стандартного розчину вихідної речовини, додають індикатор інші реактиви і титрують робочим розчином реагенту з бюретки. Відмічають об’єм робочого розчину, витраченого на титрування. Розраховують точну нормальну концентрацію (молярну концентрацію речовини еквівалента), титр робочого розчину.
Ііі. Титриметричне визначення досліджуваної речовини
В конічну колбу для титрування вміщують аліквотний об’єм досліджуваного розчину, індикатор (інші реактиви), з бюретки додають робочий розчин реагенту (титрують), відмічають об’єм реагенту, що був витрачений на титрування. За результатами титрування обчислюють нормальну концентрацію (молярну концентрацію речовини еквівалента), масу або масову частку досліджуваної речовини в розчині.
(належать
всі об’ємні визначення, в основі яких
лежить реакція нейтралізації: Н++ОН-→Н2О) При
титруванні відбувається зміна рН
розчину (зміна забарвлення). Індикатори:
лакмус, метиловий оранжевий, фенолфталеїн,
метиловий червоний, метиловий нейтральний. Стандартні
розчини:
бура Nа2В4О7
х 10Н2О,
щавлева кислота Н2С2О4
2Н2О
– готуються безпосередньо з наважок. Робочі
розчини:
НСІ, Н2SO4,
HNO3,
KOH, NaOH – готують приблизної концентрації,
точну нормальність встановлюють
титруванням, стандартних розчинів. Використовують
метод нейтралізації для визначення: кислотності
розчинів (розсолів, соків, молока),
кислотності грунтів, тіста; карбонатної
твердості води; вмісту
сірчаної кислоти в технічній H2SO4; вмісту
аміаку, білків в харчових подуктах,
лугів.Метод нейтралізації (кислотно-основного титрування)

Метод
окислювально-відновний
(редоксиметрія) (реакції
окислення – відновлення) Перманганатометрія; Йодометрія.Класифікація методів об’ємного аналізу (залежно від типу хімічної реакції)



Метод
осадження (реакції
осадження іонів у вигляді погано
розчинних сполук) Аргентометрія
(Метод Мора); Роданідометрія
(Метод Фольгарда).
Метод
комплексоутворення (реакції
зв’язування іонів в малодисоційовані
комплексні сполуки) Комплексонометрія.
Індикатори методу нейтралізації – слабкі кислоти або основи, молекулярна форма яких має інше забарвлення, ніж іонна. Колір індикатора залежить від концентрації водневих іонів розчину. Зміна кольору індикатора – результат внутрішньомолекулярного перегрупування, що спричиняє зміну будови молекули індикатора.
Інтервал кислотності розчину, в межах якого відбувається помітна зміна забарвлення індикатора, називається інтервалом переходу забарвлення індикатора (ІП).
У межах інтервалу переходу індикатора відбувається поступова зміна забарвлення.
Концентрація катіонів Н+, при якій відбувається найбільш різка зміна забарвлення, називається показником титрування (рТ). При нейтралізації значення рТ знаходиться приблизно в середині інтервалу переходу забарвлення. Титрування проводять доти індикатор не змінить свого забарвлення.
Якщо індикатор двоколірний (метиловий оранжевий), то величина рТ індикатора мало залежить від концентрації індикатора (зміна забарвлення обумовлена різкою зміною співвідношення молекулярної та іонної форм індикатора, що мають різний колір. Якщо індикатор одноколірний (фенолфталеїн), то рТ та інтервал переходу залежать від концентрації індикатора (молекулярна – безбарвна, іонна – червоний колір), для фіксації точки еквівалентності – 2-3 краплі індикатора.
Умовою правильного визначення точки еквівалентності є якнайменша різниця значень рТ вибраного індикатора і рН в точці еквівалентності.
Інтервали переходу забарвлення та показники титрування індикаторів
Назва індикатора |
Інтервал переходу, ІП |
Показник титрування, рТ |
Метиловий оранжевий |
3,1 – 4,4 |
3,8 |
Метиловий червоний |
4,4 – 6,2 |
5,4 |
Нейтральний червоний |
6,8 – 8,0 |
7,0 |
Лакмус |
5,0 – 8,0 |
6,5 |
Фенолфталеїн |
8,2 – 10,00 |
9,1 |
Висновок: для правильного вибору індикатора, необхідно:
скласти рівняння хімічної реакції, що відбувається при титруванні;
визначити рН точки еквівалентності, виходячи з продукту реакції;
порівняти рН точки еквівалентності з рТ різних індикаторів та вибрати найближче за значенням.
Вимоги до індикаторів кислотно-основного титрування:
видиме забарвлення індикатора за невеликої кількості його;
різка зміна забарвлення індикатора в невеликому інтервалі значень рН;
оборотна зміна забарвлення індикатора.
Умови титрування:
титрують завжди з одним і тим же індикатором;
для титрування беруть завжди одну й ту ж кількість крапель індикатора;
титрують завжди до одного й того ж кольору індикатора в усіх паралельних дослідах;
дотримуються одного й того ж порядку титрування (доцільно титрувати від основи до кислоти).
Випадки титрування в методі нейтралізації
Процес титрування можна передати кількісно – криві титрування, які відображають характер зміни рН середовища титрованого розчину при додаванні до нього стандартного розчину титранту (кислоти або основи).
|
|
Реакція між сильною кислотою і лугом: НСІ + NaОН → NaСІ + Н2О в точці еквівалентності рН = 7 (утворюється сіль сильної кислоти та сильної основи), середовище нейтральне. Індикатор – нейтральний, червоний (рТ = 7), лакмус (рТ = 6,5). Титрування необхідно закінчити в нейтральному середовищі. |
|
|
|
|
Реакція між сильною кислотою і слабкою основою: HCl + NH4OH → NH4Cl + H2O в точці еквівалентності утворюється сіль NH4Cl (сильної кислоти та слабкої основи). рН<5,3; кисле середовище. Титрування необхідно закінчити в кислому середовищі, індикатор – метиловий червоний (рТ=5,4). |
|
|
|
|
Реакція між слабкою кислотою і сильною основою: СН3СООН + NaOH → CH3COONa + H2O в точці еквівалентності утворюється сіль CH3COONa (слабкої кислоти та сильної основи). рН=8,7; лужне середовище. Титрування необхідно закінчити в лужному середовищі, індикатор – фенолфталеїн (рТ=9,1). |
|
|



Лекція №16. Методи окислення – відновлення
Основні питання:
Сутність окислювально-відновних методів, їх значення в проведенні хіміко-технологічного контролю.
Перманганатометрія, її суть.
Йодометрія, її суть.
Метод окислення-відновлення базується на окислювально-відновних реакціях, які відбуваються між стандартним розчином і досліджуваною речовиною.

Переваги методів окисно-відновного титрування:
велика точність, відтворюваність, простота, експресність аналізу;
можна проводити титрування різними методами(індикаторними, без індикаторними, інструментальними);
можна диференційовано визначати вміст різних компонентів у суміші, сплавах.
Особливість окисно-відновних реакцій в титриметрії:
взаємодіють не лише окисник і відновник, а й молекули середовища;
багатостадійність процесів окислення – відновлення;
швидкість окисно-відновних реакцій значно менша, ніж реакцій обміну;
при перебігу окисно-відновної реакції утворюються речовини – проміжні продукти, які часто змінюють хід самої окисно-відновної реакції;
більшість окисно-відновних реакцій – оборотні.
Вимоги щодо окислювально-відновних реакцій:
Для проведення хімічного аналізу придатні реакції, що:
відбуваються швидко, стехіометрично, до кінця (необоротні);
утворюють продукти певного хімічного складу;
дозволяють точно фіксувати точку еквівалентності;
не взаємодіють з побічними продуктами.

При аналізі використовують:
пряме титрування – при визначенні відновників. (Розчин FeSO4 титрують безпосередньо розчином КМnО4 в кислому середовищі в перманганатометрії).
обернене титрування або титрування по залишку – при визначенні відновників. (До розчину Na2SО3 додають точний об’єм титрованого розчину йоду в надлишку. Після закінчення реакції залишок йоду відтитровують стандартним розчином Na2S2O3 ∙ 5H2O).
обернене титрування замісника – при визначенні окисників. (До розчину CuSO4 додають надлишок КІ, утворений І2 (замісник) відтитровують розчином Na2S2O3).
Вимоги до індикаторів у окисно-відновному титруванні
різне забарвлення окисленої і відновленої форм;
стійкість до зовнішніх факторів (СО2, О2, світла);
видима зміна забарвлення індикатора при невеликій його кількості;
зміна кольору індикатора повинна співпадати з точкою еквівалентності;
індикатор повинен реагувати в точці еквівалентності з невеликим надлишком окисника або відновника і мати вузький інтервал дії індикатора.
Шляхи збільшення швидкості окисно-відновної реакції
зміна рН у розчині, що титрують;
підвищення температури;
використання каталізаторів;
зміна молярної концентрації реагуючих речовин.
Індикатори методу окислення-відновлення (для фіксації точки еквівалентності):
Робочий розчин є індикатором. В перманганатометрії робочий розчин KMnO4 є індикатором: при додаванні надлишку KMnO4 відбувається забарвлення розчину в рожевий колір.
Специфічні індикатори – речовини, що утворюють забарвлені сполуки з одним компонентом реакції. Крохмаль з йодом утворює сполуку інтенсивно-синього забарвлення, використовується в йодометрії.
Органічні сполуки, які здатні окислюватися або відновлюватися, причому забарвлення окисленої форми відрізняється від забарвлення відновленої форми. Індикатори: дифеніламін (оборотний), метиловий оранжевий (необоротний).
Необхідно, щоб потенціал точки еквівалентності співпадав з потенціалом зміни забарвлення індикатора.
Перманганатометрія – метод об’ємного аналізу, в якому робочим титрованим розчином є розчин перманганату калію (KMnO4).
Сутність методу перманганатометрії – окислення відновників розчином KMnO4.
Стандартний розчин – щавлева кислота (Н2С2О4 ∙ 2Н2);
Робочий розчин ‑ перманганат калію КМnО4;
Індикатор ‑ КМnО4.
Окислення kMnO4 в різних середовищах
Кисле середовище: KMnO4- ‑ дуже сильний окисник, відновлюється до Mn2+.
1 0FeSO4
+ 2KMnO4
+ 8H2SO4
→ 5Fe2(SO4)3
+ 2MnSO4
+ K2SO4
+ 8H2O
0FeSO4
+ 2KMnO4
+ 8H2SO4
→ 5Fe2(SO4)3
+ 2MnSO4
+ K2SO4
+ 8H2O
2Fe2+ - 2e → 2Fe3+ 5 окислення
10
Mn7+ + 5e → Mn2+ 2 відновлення
fекв(KMnO4)=1/5;
М(1/5
KMnO4)=![]() 31,61г/моль
31,61г/моль
Е0 MnO4 / Mn2+ = 1,51 В (стандартний окислювально-відновний потенціал).
MnO4- + 8Н+ + 5е → Mn2+ + 4Н2О
Нейтральне або слабколужне середовище:
MnO4- + 2Н2О + 3е → MnО2↓ + 4ОН-
3FeSO4 + KMnO4 + 2H2O + 5КОН → 3Fe(ОН)3↓ + MnO2↓ + 3K2SO4
F e2+ - 1e → Fe3+ 3 окислення
3
Mn7+ + 3e → Mn4+ 1 відновлення
fекв(KMnO4)=1/3;
М(1/3 KMnO4)=![]() 52,68г/моль
52,68г/моль
Е0 MnO4- / MnО2 = 0,59 В (стандартний окислювально-відновний потенціал).
Лужне середовище: MnO4- +1е → MnO42-
FeSO4 + KMnO4 + 3КОН → Fe(ОН)3↓ + КМnO4 + K2SO4
F e2+ ‑ 1e → Fe3+ 1 окислення
1
Mn7+ + 1e → Mn6+ 1 відновлення
fекв(KMnO4)=1;
М(1/5 KMnO4)=![]() 158,04г/моль
158,04г/моль
Е0 MnO4- / MnО42- = 0,56 В (стандартний окислювально-відновний потенціал).
Висновок:
Окислення KMnO4 найкраще проводити в кислому середовищі:
окислювальна здатність MnO4- в Н+ середовищі значно вища, ніж Н2О або ОН-;
знебарвлення рожевого розчину (KMnO4) дозволяє зафіксувати точку еквівалентності (кінець титрування);
індикатором є робочий розчин KMnO4 (надлишкова крапля KMnO4 забарвлює розчин в блідо-рожевий колір);
не утворюється осад MnО2, тобто не відбуваються побічні реакції.
Для створення кислого середовища використовують тільки Н2SO4 .
HNO3 – сильний окислювач, частково окислюватиме досліджуваний відновник (витрата KMnO4 буде меншою).
НСІ – відновник, окислюватиметься розчином KMnO4 (витрата KMnO4 буде більшою).
Застосування методу перманганатометрії:
Для визначення вмісту відновників (Fe2+ в питній воді) – прямим титруванням.
Для визначення вмісту Са2+, дубільних речовин, щавлевої кислоти, нітритів.
Хімічні показники якості води. Окиснюваність води
Окиснюваність визначає загальний вміст у воді відновників – органічних і неорганічних , які реагують з окисниками. У природних і стічних водах переважають органічні відновники, тому всю величину окиснюваності відносять до органічних домішок води.
Величина окиснюваності води виражається витратою окисника (КМnО4, К2Сr2О7) або еквівалентної кількості кисню, яка необхідна для окислення органічних сполук, що містяться у воді об’ємом 1 дм3.
Найменша окислюваність в артезіанській воді (до 2 мг/дм О2), в річковій воді і воді водосховищ коливається в межах від 1 до 10 мг/дм3, досягаючи більш високих показників у водах болотного походження, які місять багато гумінових речовин. Підвищена окиснюваність води свідчить про забруднення джерела виробничими або побутовими стічними водами і потребує застосування відповідних заходів по її санітарній охороні.
Окиснюваність води є оцінкою її якості, як показника забруднення органічними і неорганічними сполуками; чим вища окиснюваність води, тим гірша її якість.
Розрізняють окиснюваність:
Хімічна (використовують хімічний окисник: KMnO4, K2Cr2O7, KIO3) – називається хімічною потребою в кисні (ХПК);
Біохімічна (використовують як окисники аеробні бактерії) – називається біохімічна потреба в кисні (БПК).
Для визначення якості води за її окиснюваністю на практиці досить часто користуються орієнтовним способом.
Для цього у пробірку наливають близько 10 см3 досліджуваної води, додають до неї з краплі розчину KMnO4 з Сн(KMnO4) = 0,03 моль/л і залишають на 20 хв за кімнатної температури. Малиновий колір свідчить, що вода містить мало органічних домішок і вважається цілком задовільною; якщо забарвлення набуває червонуватого кольору – вода забруднена органічними речовинами; якщо вода має жовто-бурий колір, то така вода вважається непридатною для харчових і питних цілей.
Етапи перманганатометричного визначення
І. Приготування стандартних і робочих розчинів
Вихідна речовина: щавлева кислота Н2С2О4 ∙ 2Н2О – зважується точно на аналітичних терезах, Na2С2О4 - оксалат натрію.
Робочий розчин KMnO4: готується приблизної концентрації, наважка зважується на технохімічних терезах.
ІІ. Установлення титру та нормальності робочого розчину KMnO4 за щавлевою кислотою титруванням
Хімізм процесу:
2KMnO4 + 5H2С2O4 + 3Н2SО4 → 2MnSO4 + K2SO4 + 10СО2 + 8Н2О
С 2О42- - 2е → 2СО2 5
10
Mn7+ +5е → Mn2+ 2
5С2О42- + 2Mn7+ → 10СО2 + 2Mn2+
В конічну колбу: 20мл розчину Н2С2О4 + Н2SО4(концентрована) → нагрівання майже до кипіння
В бюретку: розчин KMnO4.
Титрування проводять з гарячого розчину: перші порції KMnO4 додають повільно до знебарвлення розчину в колбі. Титрують до появи блідо-рожевого забарвлення (надлишкова крапля KMnO4), що не зникає протягом 1хв.
ІІІ. Визначення відновника (Fe2+ в солі Мора) – титруванням
1 0FeSO4 + 2KMnO4 + 8H2SO4 → 5Fe2(SO4)3 + 2MnSO4 + K2SO4 + 8H2O
2Fe2+ - 2e → 2Fe3+ 5 окислення
10
Mn7+ + 5e → Mn2+ 2 відновлення
В конічну колбу: 20мл розчину солі Мора мірною піпеткою.
В бюретку: розчин KMnO4.
Титрують до появи блідо-рожевого забарвлення (надлишкова крапля KMnO4), що не зникає протягом 1хв.
Йодометричний метод аналізу базується на реакціях окислення-відновлення вільного йоду в йодид, або навпаки:
І20 + 2е → 2І- - йод виступає в якості окислювача (процес відновлення);
2І- - 2е → І20 – йодид-іон виступає як відновник (процес окислення);
Метод йодометрії використовують для кількісного визначення окисників і відновників.
Робочі розчини: Na2S2O3 – тіосульфат натрію, І2 – йод.
Індикатор: крохмаль, який з вільним йодом, утворює сполуку темно-синього кольору. Реакція йоду з крохмалем дуже чутлива, що обумовлює значну точність аналізу.
Стандартні розчини (вихідні речовини): K2Cr2O7 – біхромат калію, KMnO4 – перманганат калію.
Визначення нормальності та тиру розчину Na2S2O3 проводять по K2Cr2O7.
Відбувається реакція: K2Cr2O7 + 6КІ + 7H2SO4 → Cr2(SO4)3 + 3I2 + 4K2SO4 + 7H2O
2 Cr+6 + 6е → 2Cr+3 1
6
2І- - 2е → І2о 3
fекв(K2Cr2O7)=1/6;
М (1/6 K2Cr2O7)=![]() 49г/моль
49г/моль
2Cr+6 + 6I- → 2Cr+3 + 3I20
Вільний йод, що виділився, титрують розчином тіосульфату натрію у присутності крохмалю до зникнення синього забарвлення розчину (розчин набуває зеленого кольору із-за наявності Cr+3).
Визначення окисників проводять методом оберненого титрування замісника (по заміщенню):
Розчин окисника + надлишок КІ + надлишок H2SO4 → виділення І2 (буре забарвлення розчину) → титрують розчином Na2S2O3 до появи жовтого забарвлення в кінці титрування додають крохмаль (поява синього забарвлення) → титрують Na2S2O3 до зникнення синього кольору.
Хімізм процесу:
10КІ + 2KMnO4 + 8H2SO4 → 5I2 + 2MnSO4 + 6K2SO4 + 8H2O
2 І- ‑ 2е → І20 5
10
Mn+7 + 5е → Mn+2 2
Сумарна реакція: 10І- + 2Mn+7 → 5І20 + 2Mn+2
І2 + 2Na2S2O3 → 2NaI + Na2S4O6
Визначають: KMnO4, КІО3, AlCl3, CuSO4.
Визначення відновників проводять методом оберненого титрування (по залишку)
Розчин відновника + надлишок робочого розчину І2 → суміш витримують деякий час → надлишок І2 титрують робочим розчином Na2S2O3 у присутності крохмалю, який додають до досліджуваного розчину перед титруванням.
Хімізм: SnCl2 + І2 + 2НСІ → SnCl4 + 2НІ
Визначають: тіосульфати, H2S, SnCl2, Fe2+.
Умови проведення йодометричних визначень
Титрувати на холоді (запобігання випаровуванню йоду).
Оптимальне значення рН=5-6 (в сильно лужному середовищі І2 реагує з лугом), а в сильно кислому середовищі І2 реагує з кислотою).
Надлишок КІ (прискорюється реакція між КІ та окисником, розчиняє І2).
Реакції між окисником та КІ відбуваються недостатньо швидко, тому титрують йод, що виділився, через 3-4 хвилини.
Реакцію бажано проводити в темноті, щоб запобігти взаємодії КІ з киснем повітря.
При визначенні відновників (титрування І2), крохмаль додають до досліджуваного розчину перед титруванням, при визначенні окисників, кислот – крохмаль додають в кінці титрування.
Лекція № 17. Метод осадження
Основні питання:
Сутність методу осадження.
Метод Мора.
В основі об’ємно-аналітичних методів осадження лежать реакції, в результаті яких відбувається утворення погано розчинних сполук, тобто реакції осадження.
NaCl + AgNO3 → AgCl↓ + NaNO3
AgNO3 + KSCN → AgSCN↓ + KNO3
Вимоги щодо реакцій осадження:
Осад повинен бути практично нерозчинним (>10-6моль/л).
Миттєве утворення осаду від час титрування.
Відсутність адсорбції реагуючих речовин осадом.
Можливість фіксації точки еквівалентності.
Прийоми проведення титрування:
Пряме титрування (в нейтральному середовищі) по Методу Мора.
Обернене титрування (в кислому середовищі) за методом Фольгарда. Спочатку кількісно осаджують хлорид-іони додаванням надлишку катіонів аргентуму. Потім надлишок катіонів аргентуму відтитровують роданід-іонами.
Застосування методів осадження:
для визначення хлоридів методом Мора (в нейтральному середовищі);
визначення срібла у сплавах (сплав розчиняють в азотній кислоті, утворений розчин титрують розчином роданіду амонію у присутності залізо амонійного галуну як індикатора);
для аналізу солей срібла, роданідів, бромідів, хлоридів, йодидів методом Фольгарда (в нейтральному та кислому середовищі).


Методи осадження

Хімізм методу Мора:
NaCl + AgNO3 → AgCl↓ + NaNO3
Ag+ + Cl- → AgCl↓
Утворення AgCl – осаду білого кольору – відбувається доти в розчині містяться СІ-.
K2CrO4 + 2AgNO3 → Ag2CrO4↓ + NaNO3
2Ag+ + CrO42- → Ag2CrO4↓
Ag2CrO4 – осад цегляно-червоного кольору, з’являється тільки після практичного повного осадження СІ- у вигляді білого осаду AgCl (розчинність Ag2CrO4 більша ніж розчинність AgCl).
В точці еквівалентності додавання однієї краплі розчину AgNO3 викликає утворення цегляно-червоного осаду Ag2CrO4.
Умови проведення визначення за методом Мора:
Концентрація індикатора K2CrO4 = 10-2моль/л
Надлишок K2CrO4 сприяє передчасному утворенню Ag2CrO4 (розчин недотитрований).
Нестача K2CrO4 викликає утворення Ag2CrO4 при надлишку Ag+ (розчин перетитрований).
Титрування необхідно проводити в нейтральному середовищі.
В кислому середовищі осад Ag2CrO4 не утворюється (добре розчинений в кислотах):
Ag2CrO4↓ + 2HNO3 → 2AgNO3 + H2CrO4
Ag2CrO4↓ + 2H+ → 2Ag+ + CrO42-
В лужному середовищі осад Ag2CrO4 не утворюється, а осадження Ag2O (бурий) на початку титрування заважає визначенню точки еквівалентності:
2AgNO3 + 2NaOH → Ag2O↓ + H2O + 2NaNO3
2Ag+ + 2OH- → Ag2O↓ + H2O
В аміачному середовищі СІ- та CrO42- розчиняється з утворенням комплексних аміакатів срібла:
Ag2CrO4↓ + 4NH4OH → [Ag(NH3)2]CrO4 + 4H2O
AgСІ + 2NH4OH → [Ag(NH3)2]СІ + 2H2O
Заважають іони: СО32-, РО43-, С2О42-, SO32-, S2- (утворюють з Ag+ осади), Fe3+, Al3+ (підлягають гідролізу), Ba2+, Sr2+, Pb2+, Bi3+ (утворюють осади з K2CrO4), Ni2+, Co2+, Cu2+ (забарвлені).
Схема проведення аналізу
В бюретку: розчин AgNO3;
В конічну колбу: аліквотний об’єм досліджуваного розчину (NaCl) + розчин K2CrO4 (1-2 краплі насиченого);
Титрування: поява білого осаду (АgСl);
Кінець титрування: поява цегляно-червоного осаду Ag2CrO4.
Лекція № 18. Метод комплексонометрії
Основні питання:
Сутність методу комплексонометріїї.
Застосування методу комплексонометрії.
Комплексонометрія – метод, що базується на реакції утворення міцних комплексних сполук іонів металів з органічними реактивами (комплексони). Найбільш поширені комплексони: ЕДТА (етилендіамінтетраоцтова кислота), Na2H2У (трилон Б, комплексон ІІІ).
Хімічна формула трилону Б: Хімічна формула комплексної солі кальцію:
H

 OOCH2C–N-CH2-CH2-N-CH2COOH
O=C
‑ O
O
‑ C=O
OOCH2C–N-CH2-CH2-N-CH2COOH
O=C
‑ O
O
‑ C=O
 Ca
Ca


 NaOOC-CH2
CH2-COONa
H2C
‑ N
N
‑ CH2
NaOOC-CH2
CH2-COONa
H2C
‑ N
N
‑ CH2
NaOOC-CH2 CH2 – CH2 CH2-COONa
Зв’язування досліджуваного катіону в комплекс відбувається тим краще, чим міцніший утворений комплекс.
Переваги методу комплексонометрії:
Утворення міцних сполук комплексонів з катіонами металів (крім Na+, K+, Li+) у співвідношенні 1:1.
Утворення стійких комплексів з катіонами лужноземельних металів – Mg2+, Ca2+, Ba2+, Sr2+.
Висока чутливість (до 10-3моль/л).
Реакції відбуваються швидко та стехіометрично.
Висока селективність.
Можливість визначення одних металів у присутності інших, регулюючи кислотність середовища:
рН<7 : Fe3+, Al3+ (використовують буферну суміш СН3СООН + СН3СООNa);
рН>7 : Ba2+, Ca2+, Mg2+ (використовують буферну суміш NH4Cl + NH4OH).
Стандартний розчин: трилон Б (готують з точної наважки) або використовують фіксанал.
Хімічна формула трилону Б: Na2H2C10H12O8N2 ∙ 2H2O, молярна маса 327,30г/моль, fекв=1/2;
М(1/2
трилону Б) =
![]() =186,15г/моль
=186,15г/моль
Молярну концентрацію речовини еквівалента, титр розчину комплексону ІІІ можна встановити за MgSO4, розчин якого готують з фіксаналу.
Індикатори (дозволяють зафіксувати точку еквівалентності), застосовують:
еріохром чорний Т;
кислотний хром темно-синій (синій колір, комплексні сполуки з металами – вишнево-червоні);
мурексид (темно-червоний, водний розчин – фіолетово-червоний залежно від рН, комплексні сполуки – червоного кольору).
Металіндикатори – органічні барвники, які утворюють з катіонами металу розчинні у воді забарвлені комплексні сполуки, причому комплексі сполуки індикатора з катіонами металу менш міцні ніж внутрішньокомплексні солі металу з комплексоном.
Еріохром чорний Т стійкий в твердому вигляді, для визначення твердості води використовують суміш індикатора з хлоридом натрію або калію в співвідношенні 1:200. Для кожного титрування беруть 20-30 мг суміші на 100мл досліджуваного розчину.
При титруванні в точці еквівалентності спостерігається зміна забарвлення за рахунок руйнування комплексної сполуки індикатора з катіоном металу та виділення індикатора у вільному стані.
Аміачний буферний розчин готують таким чином: у мірну колбу на 1л вносять 67,5г хлориду амонію, доливають 200мл дистильованої води і ретельно перемішують до повного розчинення солі. Потім додають 570мл концентрованого розчину гідроксиду амонію, доводять об'єм до помітки дистильованою водою і перемішують. рН одержаного розчину дорівнює 10.
Застосування методу комплексонометрії:
Для визначення загальної твердості води (Са2+ + Mg2+).
Для визначення Fe3+, Al3+, Se3+, Zn2+, Zr2+, Co2+, Cu2+ та ін. (аналіз різних сплавів, мінералів, силікатних матеріалів).
Хімічні процеси, що відбуваються в розчині, при визначенні Са2+ трилонометричним методом
Досліджуваний розчин (Са2+) + буферний розчин (NH4Cl+NH4OH) + індикатор еріохром чорний Т → розчин вишнево-червоного кольору (утворення комплексної сполуки Са2+ з ЭХЧ-Т): Са2+ + H2Ind ↔ CaInd + 2H+ (ЭХЧ-Т) вишнево-червоний розчин.
При титруванні трилон Б зв’язує Са2+ в більш міцний комплекс (безбарвний):
CaInd + Na2H2Y → Na2CaY + H2Ind
вишн.-черв.розч. безбарвний трилон Б безбарвний синій (ЭХЧ-Т)
В результаті титрування виділяється вільний ЭХЧ-Т, який при рН=8-9 забарвлений в синій колір.
Кінець титрування: зміна забарвлення з вишнево-червоного на синє.
В природі вода ніколи не зустрічається у вигляді чистої сполуки. Маючи властивості універсального розчинника, вона містить значну кількість різних речовин, вміст та співвідношення між якими визначаються умовами формування води, складом водоносних порід, залежно від цих та інших умов вода має різний хімічний та мікробіологічний склад та різні органолептичні властивості.
Твердість води обумовлена наявністю іонів кальцію та магнію. Іони кальцію надходять у воду при розчиненні вапняків чи гіпсу. Основне джерело надходження іонів магнію – доломіти та продукти вивітрювання гірських порід.
Хімічним показником , що характеризує твердість води, є сумарний вміст мг-еквівалентів іонів кальцію та магнію в 1л води.
Загальна твердість води складається з тимчасової та постійної твердості. Тимчасова твердість обумовлена вмістом гідрокарбонатів кальцію та магнію. Під постійною твердістю розуміють вміст у воді інших солей кальцію та магнію.
Показник твердості води аналізується згідно таких нормативних документів:
ГОСТ 2874 „Вода питьевая. Гигиенические требования и контроль за качеством”;
ДСанПіН “Вода питна. Гігієнічні вимоги до якості води централізованого господарсько – питного водопостачання” від 15.04.97р. за № 136/1940.
Показник якості (загальна твердість води) визначається комплексонометричним методом, який регламентований ГОСТ 4151 „Вода питьевая. Метод определения общей жёсткости”.
Лекція №19. Фізико-хімічні методи аналізу. Фотометричний метод аналізу
Основні питання:
Класифікація фізико-хімічних методів. Практичні напрямки використання фізико-хімічних методів аналізу.
Фотометричні методи аналізу, їх сутність:
інструментальні методи (фотоелектроколориметрія). Апаратура фотометричного методу аналізу;
методи візуальної колориметрії.
Фізико-хімічні методи аналізу – це методи аналізу, що базуються на залежності фізичної характеристики речовин (світлопоглинання, світлозаломлення, електричної провідності, теплопровідності тощо) від їх хімічного складу. Інтенсивність фізичного сигналу залежить від концентрації досліджуваного компоненту. Виміри проводять за допомогою відповідної апаратури (інструментальні методи).
Аналітичний сигнал – будь який прояв хімічних та фізичних властивостей об’єкта аналізу, який можна використати для визначення кількісної та якісної оцінки компонентів речовини.
Фізико-хімічні методи аналізу (інструментальні методи) – окремий розділ аналітичної хімії, завданням якого є визначення кількісного та якісного складу речовин при зміні їхніх фізико-хімічних властивостей.
Переваги фізико-хімічних методів аналізу:
швидкість визначення;
висока чутливість; висока вибірковість;
можливість безперервного виконання аналізів;
;можливість широкого застосування комп’ютерних баз даних і програмного забезпечення.
Задачі, що розв’язуються за допомогою фізико-хімічних методів аналізу:
Визначення якості сировини.
Контроль процесів виробництва.
Аналіз відходів виробництва з метою їх утилізації та охорона навколишнього середовища.
.Контроль якості виробленої продукції.
Визначення сировини та в продовольчих і непродовольчих товарах гранично допустимих концентрацій (ГДК) токсичних речовин (Нg, Pb, Cd, канцерогенні сполуки, ароматичні вуглеводні, N- нітрозоаміни).
Місце фхма серед хімічних та фізичних методів дослідження

Роль фізико-хімічних методів аналізу для сертифікації продукції
Зараз в Україні введено єдиний реєстр сертифікації харчових продуктів, відповідність якому визначається за допомогою фізико-хімічних методів аналізу. Кожному продовольчому товару надається спеціальний сертифікат відповідності, який має 12 ступенів захисту. Введення цього сертифіката регламентується законом захисту прав споживачів України. Кожний продовольчий товар повинен мати на етикетці знак відповідності:
1. |
|
‑ продукт відповідає всім вимогам нормативних документів |
2. |
|
‑ продукт відповідає всім обов’язковим вимогам нормативних документів |
3. |
|
‑ продукт відповідає окремим вимогам нормативних документів |
Сертифікат відповідності призначено для:
Запобігання розповсюдження недоброякісної продукції;
Власного висновку споживача про ступінь якості товару.
Спектроскопічні методи аналізу базуються на взаємодії речовин з електромагнітним випромінюванням.
Фотометрія, спектрометрія дають змогу визначити практично всі елементи в навколишньому середовищі, а також у виробах промислової та харчової галузі:
Емісійна фотометрія – для визначення лужних і лужноземельних елементів – високочутлива і проста у використання;
Атомно адсорбційний аналіз – для визначення мікроелементів;
Рентгенофлуоресцентний аналіз – перспективний для визначення всіх елементів Періодичної системи (потребує складної і дорогої апаратури);
Інфрачервона (ІЧ) спектрометрія – для визначення структури органічних речовин, полімерних матеріалів, аналізу біологічних об’єктів.
Електрохімічні методи ґрунтуються на електрохімічних властивостях досліджуваних речовин:
Пряма потенціометрія (іонометрія) використовується для визначення іонів К+, Na+,Ca2+, NO3-, Br-, CI- у воді, а також визначення рН у сертифікації харчових продуктів;
Вольтамперометрія використовується для визначення мікроелементів, а також для дослідження взаємодії елементів з органічними речовинами;
Кондуктометрія використовується для визначення загального вмісту солей у водних і неводних розчинах;
Кулонометрія використовується для визначення вмісту важких металів.
Хроматографічні методи базуються на сорбційних процесах і використовуються для розділення та ідентифікації мікро кількостей органічних і неорганічних речовин:
Газорідинна хроматографія – для аналізу складних сумішей органічних речовин;
Розподільча хроматографія на папері в тонкому шарі – для аналізу мікрокількостей амінокислот, спиртів, вуглеводнів.
Іонообмінна хроматографія – для розділення органічних і неорганічних речовин.






Фотометричний аналіз (молекулярна фотоабсорбційна спектроскопія) належить до оптичних методів аналізу. Ґрунтується на здатності речовин поглинати електромагнітне випромінювання оптичної частини спектра.
Спектр поглинання (спектральна характеристика речовини) – залежність світлопоглинання (А, Т, ε або їх логарифмів) від характеристики падаючого світла.
Електромагнітне випромінювання характеризується такими параметрами:
Довжина хвилі λ (відстань, яку проходить хвиля за один період коливань, мкм, нм);
Частота випромінювання υ (частота коливань за секунду, Гц);
Енергія випромінювання Е, вимірюється в електронвольтах (еВ)
Е = h∙υ, де h – постійна Планка (6,626916∙10-34Дж∙с).
Оптичний спектр містить ультрафіолетову (УФ), видиму та інфрачервону (ІЧ) області.
Спектральна ділянка |
Довжина хвилі λ, нм |
Енергія, еВ |
Процеси, що відбуваються в результаті поглинання (випромінювання) світла |
Ультрафіолетова: вакуумна |
< 200 |
102 ‑ 10 |
Електронні переходи |
близька |
200 ‑ 400 |
||
Видима |
400 ‑ 700 |
10 ‑ 1 |
Електронні переходи |
Інфрачервона: близька |
700 – 1,5∙103 |
1 – 10-2 |
Коливання молекул |
фундаментальна |
1,5∙103 – 7,5∙104 |
Обертання молекул |
|
далека |
7,5∙104 ‑ 106 |
Основою фотометричного методу аналізу є вибіркове поглинання світла частинками (молекулами або іонами) речовини у розчині.
При поглинанні світла змінюється внутрішня енергія молекули речовини, молекула переходить на більш високий енергетичний рівень. Кожна молекула має певний набір збуджених квантових станів, які відрізняються за значенням енергії і тому інтенсивно поглинають ті кванти, енергія яких дорівнює енергії збудження молекули.
Характер поглинання залежить від природи речовини, на цьому будується якісний аналіз, а на залежності світлопоглинання від концентрації ґрунтується кількісний аналіз речовин.
Колориметричні (фотометричні) методи аналізу базуються на реакціях, що супроводжуються утворенням забарвлених розчинних сполук (іноді руйнуванням забарвлених сполук).
Переваги колориметричних методів аналізу:
Можливість визначення дуже малих кількостей елементів (10-4-10-5М); 1 крапля (0,05мг) KMnO4 в 100мл розчину.
Швидкість визначення, простота методики.
Можливість використання хімічних реакцій різних типів.
Недоліки колориметричних методів: реакції малоспецифічні.
Вимоги щодо реакцій в колориметрії:
повинні проходити достатньо швидко з утворенням малодисоційованої речовини;
достатньо інтенсивне забарвлення продукту реакції;
стійкість у часі забарвленої сполуки;
постійний склад забарвленої речовини.
Типи хімічних реакцій в колориметрії:
Реакції утворення забарвлених неорганічних комплексних сполук: аміакатів, галогенідних комплексів, пероксидних сполук, гетерополікислот.
Реакції утворення забарвлених комплексів з органічними реагентами: сполуки алюмінію з алізарином S; кобальту з
 -натрозо-
-натрозо- нафтолом;
магнію з титановим жовтим.
нафтолом;
магнію з титановим жовтим.Реакції окислення – відновлення з утворенням забарвлених сполук: окислення Mn2+ до Mn7+, утворений MnO4 - має інтенсивне рожево-фіолетове забарвлення.
Вимірюють інтенсивність забарвлення досліджуваної речовини в розчині (NiSO4, CuSO4).
Етапи колориметричного (фотоелектроколориметричного) визначення
І етап. Досліджуваний компонент за допомогою хімічної реакції переводять в забарвлену сполуку
X + R → П, де Х – досліджуваний компонент;
R – реагент;
П – продукт (забарвлена сполука).
Забарвлення будь-якого розчину характеризується спектром поглинання. Про кількість досліджуваного компоненту судять за інтенсивністю забарвлення розчину (чим інтенсивніше забарвлення, тим більша концентрація елемента в розчині).
ІІ етап. Вимірюють інтенсивність забарвлення розчину
Забарвлення досліджуваного розчину порівнюють із забарвленням стандартного розчину (з точно визначеною концентрацією досліджуваного компоненту). При проходженні світла крізь поглинальний розчин інтенсивність світла зменшується, що підпорядковується закону Бугера – Ламберта – Бера.
Інтенсивність забарвлення пов’язана з інтенсивністю поглинання світла.
Інтенсивність поглинання світла залежить від:
концентрації забарвленої сполуки в розчині;
товщини поглинаючого шару.
lg![]() =E
=E
![]() L
× C,
де
L
× C,
де
І0 – інтенсивність потоку світла, що входить в розчин;
Іt – інтенсивність світла, що пройшов через розчин;
L – товщина поглинаючого шару;
lg ‑ оптична густина – характеризує ступінь ослаблення (поглинання) світла при проходженні його через розчин. Позначається через А.
С – концентрація забарвленої сполуки в розчині (моль/л);
Е – молярний коефіцієнт поглинання („Епсилон”).
Фізична суть Е: молярний коефіцієнт погашення чисельно дорівнює оптичній густині 1М розчину при товщині поглинаючого шару 1см. Характеризує внутрішні властивості речовини та не залежить від об’єму розчину, товщини шару, інтенсивності освітлення; є найбільш важливою характеристикою можливої чутливості фотометричного визначення.
А = Е × L × С закон Бугера – Ламберта – Бера (основний закон світлопоглинання).
Справедливий за умов: L >> λ; С – не дуже велика.
Формулювання:
Оптична густина розчинів пропорційна добутку концентрації забарвленої речовини та товщині поглинаючого шару.
При підпорядкуванні розчину закону Бугера – Ламберта – Бера спостерігається пряма пропорційна залежність
А= f(C) при постійній товщині поглинаючого шару.
Кількісне визначення речовини за світлопоглинанням ґрунтується на використанні закону Бугера – Ламберта – Бера. Оптичну густину досліджуваного розчину вимірюють за допомогою фотоелектроколориметра або спектрофотометра, а потім розрахунковим або графічним шляхом знаходять концентрацію. Останні дві операції на сучасних приладах виконують комп’ютери.
Для вимірювання світлопоглинання досліджуваного розчину вибирають довжину хвилі світлового потоку, яка відповідає максимуму смуги поглинання.
Для якісного аналізу досліджуваного компонента за світлопоглинанням вибирають світлофільтр, який пропускає випромінювання, що досліджуваний компонент поглинає, тобто мінімум поглинання світла світлофільтром повинен відповідати максимуму поглинання розчином.
Н
‑ 1
‑ 2





Методи візуальної колориметрії



(готують
1 стандартний розчин) Порівнюють
інтенсивність забарвлення досліджуваного
та стандартного розчинів шляхом
розведення водою розчину з більш
інтенсивним забарвленням. Використовують
градуйовані циліндри.
hх
– та hстанд.-
висота рідини в досліджуваному та
стандартному розчинах після розведення.
Застосовують,
якщо інтенсивності забарвлень
досліджуваного та стандартного розчинів
близькі.Метод розведення
![]() ,
де
,
де
Апаратура фотометричного методу
Прилад для вимірювання світлопоглинання повинен виконувати такі завдання:
розклад поліхроматичного світла і виділення потрібного інтервалу довжин хвиль;
вимірювання поглинання світла речовиною.
Кожний спектральний прилад включає:
джерело випромінювання;
пристрій для виділення потрібного інтервалу довжин хвиль (монохроматор або світлофільтр);
кюветне відділення;
детектор, перетворювач сигналу;
індикатор сигналу (шкала або комп’ютерний дисплей).
Залежно від досліджуваної області спектра використовують різні джерела випромінювання (ксенонова лампа, воднева лампа, вольфрамова лампа, лампа Нернста), монохроматори (кремнієва призма, скляна призма, дифракційна решітка, інтерференційні світлофільтри, скляні світлофільтри), детектори (фотопримножувачі, фотоелементи з зовнішнім фотоефектом, вентильні фотоелементи, термопара).
В фотоелектроколориметрії ступінь поглинання світла (інтенсивність забарвлення розчину) визначають за допомогою фотоелемента.
Фотоелемент ‑ металева пластинка, вкрита шаром напівпровідника (селен, сульфід свинцю). Світловий потік після потрапляння на фотоелемент, збуджує в ньому електричний струм. Величина фотоструму, що виник, прямо пропорційна світловому потоку, що падає (закон Столетова). Струм, що виник, реєструється ввімкненим у ланцюг чутливим мікроамперметром, відхилення стрілки якого прямо пропорційно освітленості фотоелемента.
Перетворення світлової енергії в електричну на фотоелементі пов’язано з явищем фотоефекта (відрив електронів від атомів різних речовин під впливом світлової енергії).
Залежно від кількості фотоелементів, що використовуються при вимірюванні, розрізняють:
фотоелектроколориметри з одним фотоелементом (однопроменеві прилади);
фотоелектроколориметр з двома фотоелементами (двопроменеві прилади). Найчастіше використовують ФЕК-М; ФЕК-56М; ФЕК-Н-57.
У фотометрах використовують світлофільтри, в спектрометрах – призми та дифракційні решітки.
Оптична схема приладу фотоелектроколориметр ФЕК-56М (мал. 4.1)
Світловий потік від джерела світла, проходить через світлофільтр, попадає на призму, яка розділяє світловий потік на два потоки: лівий та правий. Так як джерело світла находиться у фокусі лінзи, то обидва потоки відображаються від дзеркал, проходячи через лінзи, і виходять паралельно. Паралельні світлові потоки проходять через кювети (з розчином і розчинником) і потрапляють на фотоелементи. До правого світлового потоку можуть бути включені послідовно одна або друга кювети. Діафрагма, розміщена в правому потоці світла, при обертанні зв’язаного з нею барабану, змінює свою площину і тим самим змінює інтенсивність світлового потоку, що падає на правий фотоелемент. Ліва діафрагма, розміщена в лівому світловому потоці, лівий барабан, зв’язаний з діафрагмою, використовуються для послаблення інтенсивності лівого світлового потоку, що падає на лівий фотоелемент. Правий відліковий барабан є вимірювальним, має дві шкали: шкала світлопропускання (чорна) і шкала світлопоглинання (червона), а лівий барабан – компенсаційний. Електричні струми, що виникли у двох фотоелементах надходять на мікроамперметр. Схема приладу побудована таким чином, що за однакової освітленості обох фотоелементів виникають однакові фотоструми, але протилежні за напрямом (стрілка мікроамперметра не відхилятиметься від „0”). Якщо освітленість фотоелементів різна, то компенсації струмів немає, стрілка мікроамперметра відхилиться від „0”. Змінюючи розкриття діафрагм, можна зрівняти світлові потоки, що падають на обидва фотоелемента. Про рівність світлових потоків свідчить положення стрілки мікроамперметра на „0”.
Оптична схема колориметра фотоелектричного концентраційного КФК-2 (мал. 4.2)
Принцип дії приладу базується на реєстрації сили струму, що виникає на фотоелементі. Сила струму залежить від інтенсивності забарвлення розчину (чим більша концентрація забарвленої речовини, тим більше світло поглинання і менша інтенсивність світлового потоку, що виходить з розчину, менше фотострум. Для врахування втрат світла внаслідок його відбиття від стінок кювети та його поглинання розчинником вимірювання завжди проводять відносно „нульового” розчину (вода).
Використовується для вимірювання на окремих ділянках діапазону довжин хвиль 315 – 980нм, виділених світлофільтрами, коефіцієнтів пропускання та адсорбційності розчинів, а також визначення концентрації речовин у розчинах методом побудови градуйованих графіків. Дозволяє проводити вимірювання коефіцієнтів пропускання зависі, емульсій та колоїдних розчинів у світлі, що проходить.
Світло від лампи конденсором відображається у площині діафрагми і об’єктивами переноситься у площину, що знаходиться від об’єктиву на відстані 300мм. Кювета з досліджуваним розчином вводиться у світловий потік між захисними скельцями. Для виділення вузьких ділянок спектру використовуються світлофільтри. Теплозахисний світлофільтр вводиться у світловий потік при роботі у видимій області спектру (400-490нм). Для ослаблення світлового потоку при роботі в спектральному діапазоні (400 – 500нм) встановлені нейтральні світлофільтри. Світловий потік, проходячи через світлофільтри і кювету, попадає на фотоелемент і збуджує струм, який реєструється мікроамперметром.
Фотометр |
|
|
|
|
|
|
|
|
|
|
|
|
|
Кювета |
|
|
|
|
|
|
|
|
Спектрофотометр |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



Кювета з досліджуваним розчином – робоча, а кювета з розчином порівняння – кювета порівняння; обидві кювети повинні бути якомога ідентичними. Основна вимога до кювет – прозорість в області спектра, в якій проводиться вимірювання оптичної густини. Для роботи у видимій області спектра кювети виготовляють із скла, а в ультрафіолетовій – з кремнію. Кювети бувають прямокутні, термостатовані, проточні, циліндричні.
Спектрофотометрія базується на основі вимірювання поглинання монохроматичного випромінювання ультрафіолетової, видимої та інфрачервоної області спектра. Такі вимірювання проводять за допомогою спектрофотометрів. Спектрофотометри мають більш складну конструкцію і часто – електронні пристрої (підсилювачі фотоструму, дисплеї).
|
Мал.4.1.
|
|
Мал.4.2.
|


Нефелометричний і турбідиметричний методи аналізу
Нефелометричний метод аналізу ґрунтується на визначенні концентрації речовини шляхом вимірювання інтенсивності світла, розсіяного часточками суспензії або емульсії.
Турбідиметричний метод аналізу ґрунтується на вимірюванні інтенсивності світла, яке пройшло через досліджувану систему.


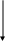 І
І
Нефелометрія. У нефелометричному аналізі інтенсивність розсіяного світла вимірюється в напрямку, перпендикулярному падаючому світловому потоку. Залежність між інтенсивністю розсіювання світла Ін і деякими параметрами системи визначається рівнянням Релея:
К
=
 Ір
= К
Ір
= К![]() ,
де І0
– інтенсивність падаючого світла;
,
де І0
– інтенсивність падаючого світла;
n1 – показник заломлення дисперсної фази;
n0 – показник заломлення дисперсійного середовища;
ν – число частинок в одиниці об’єму;
λ – довжина хвилі падаючого світла.
Ін = k ∙І0 С
Висновки:
Розсіювання світла тим більше, чим більше показник заломлення дисперсної фази n1 відрізняється від показника заломлення дисперсійного середовища n0. Якщо n1 = n0, то світло не розсіюється.
Дає змогу визначити за експериментальними даними розміри частинок, тобто їх об’єм V і радіус r, при відомій концентрації частинок ν.
Для нефелометричного аналізу спочатку будують калібрувальний графік для серії стандартних суспензій, потім нефелометрують досліджувану суспензію і за калібрувальним графіком знаходять концентрацію речовини, яку визначають.
Для вимірювання інтенсивності світла, що розсіюється, користуються нефелометрами. Для визначення концентрації колоїдних розчинів, суспензій, емульсій використовують нефелометр НФК (мал. 4.3).
 Мал..4.3.
Мал..4.3.
Турбідиметрія. Зв’язок між інтенсивністю світла, що пройшло крізь суспензію, і концентрацією частинок у зависі визначається рівнянням:
lg =k L × C, де
І0 – інтенсивність потоку світла, що входить в розчин;
Іt – інтенсивність потоку світла, що пройшов через розчин;
L – товщина поглинаючого шару;
С – концентрація частинок у зависі;
k – молярний коефіцієнт мутності.
Турбідиметричні визначення проводять у фотоелектроколориметрах або турбідиметрах.
|
|


Застосування нефелометричного і турбідиметричного методів аналізу для визначення:
Сульфат-іонів шляхом осадження їх іонами барію;
Хлорид-іонів осадженням у вигляді хлориду срібла;
Для дослідження колоїдних розчинів (мила, фарб, латексів).
Лекція №20. Хроматографічний аналіз
Основні питання:
Сутність хроматографії. Хроматографічний аналіз.
Види хроматографії:
газова, рідинна, газорідинна хроматографія;
колоночна та площинна хроматографія.
Хроматографія – метод розділення, виявлення та визначення речовин, базується на різному розподілу речовин між двома фазами, що не змішуються залежно від їх адсорбційної здатності.
1903р. – російський вчений-ботанік М.С.Цвет (розділення хлорофілу на складові компоненти).
Хроматографічний процес полягає у переміщенні рухомої фази, що містить суміш компонентів, відносно нерухомої.
Рухома фаза:
рідина (розчин досліджуваної суміші речовин);
газ (суміш газів або парів речовин).
Нерухома фаза:
тверда речовина або адсорбована рідина на твердій речовині (носій, сорбент).
Під час руху рухомої фази вздовж нерухомої компоненти адсорбуються на нерухомій фазі (сорбенті). Адсорбовані сорбентом молекули можуть перейти в рухому фазу та рухатися разом з нею далі, потім знову сорбуватися, тобто відбувається розподіл молекул кожного компоненту між двома фазами. Так як компоненти суміші мають різну спорідненість до сорбенту, то при переміщенні суміші вздовж сорбенту відбувається розподілення її на складові.
Хроматографічний процес поєднує два фактори:
термодинамічний (встановлення рівноваги між фазам);
кінетичний (рух компонентів з різною швидкістю).
Зі збільшенням спорідненості компонента до сорбенту швидкість руху компонента зменшується і він довше затримується на сорбенті.
Хроматографічний аналіз складається:
І етап. Розділення речовин.
ІІ етап. Ідентифікація та кількісне визначення речовин.
Прийоми:
Збирають фракції після хроматографування та аналізують їх за допомогою методів кількісного аналізу;
Використовують детектори для вимірювання аналітичного сигналу компонентів, що виходять з колонки та самописці для реєстрації цього сигналу.
Детектор – пристрій для реєстрації компонентів, які виходять з колонки.
за теплопровідністю (катарометр) – вимірює різницю в теплопровідності чистого газу (носія) та відокремленого компоненту суміші. Застосовують для розділення неорганічних і органічних сполук.
полум’яно-іонізаційний (спалюють газ-носій з розділеними компонентами у водневому полум’ї) – вимірює електропровідність. Застосовують для розділення органічних сполук.
електронозахватний ( -частинки іонізують газ-носій, компоненти суміші відновлюється) вимірюють зниження електропровідності. Застосовують для визначення S, P, N галогенів.
Запис сигналу детектора, як функції часу або об’єму газу-носія, відображається кривою елюювання – хроматограмою.
Р езультат
хроматографування
‑ хроматограма (крива елюювання).
езультат
хроматографування
‑ хроматограма (крива елюювання).
Хроматограма включає:
нульову лінію, яка відповідає протіканню через детектор чистого газу-носія;
час утримання (τ®) – час, необхідний для елюювання речовин до їх максимальної концентрації;
об’єм утримання (V®) – об’єм газу, необхідний для вилучення з хроматографічної колонки максимальної кількості речовини.
Основа хроматографічного аналізу – розшифрування хроматограм.
Положення піку на хроматограмі – якісний аналіз (ідентифікація речовин, ступінь чистоти сполук і поява додаткових піків свідчить про забруднення сполук).
Висота або площа піку – кількісний аналіз.
На діаграмі зображено величини, які характеризують пік кривої: А′ ‑ введення проби; А – вихід несорбувального компонента; ВДС – вихід аналізованого компонента; АС – загальний об’єм утримання Vτ, або час утримання τ; А′С – нульова лінія; АС – час утримання; h – висота піка

Колоночна
хроматографія
(розділення
компонентів суміші в хроматографічній
колонці).
Належать:
іонообмінна хроматографія, рідинна,
ВЕРХ (високоефективна рідинна
хроматографія). Площинна
хроматографія
(розділення
компонентів на папері або в тонкому
шарі адсорбенту).
Належать:
хроматографія на папері, ТШХ (тонкошарова
хроматографія).За способом проведення:

Види хроматографії


Адсорбційна
(колоночна, іонообмінна хроматографія,
ТШХ, газоадсорбційна хроматографія). Розподільна
(паперова,
газорідинна, газорозподільна
хроматографія). Гель-фільтраційна
– базується на різниці в розмірах
молекул речовин, що розділяють
(проникаюча
хроматографія).За механізмом розподілу речовин між фазами:
За
агрегатним станом суміші, яку розділяють: газова
хроматографія; газорідинна
хроматографія; рідинна
хроматографія.
Газова хроматографія використовується для розділення та визначення компонентів летких сумішей. Використовуються спеціальні прилади – хроматографи (мал.4.3), (мал.4.4).
|
|
|
|
|
|
|
|
|



Вибір газу-носія залежить від детектора, а швидкість потоку встановлюється постійною.
Суміш, що розділяють, вводять за допомогою мікрошприца.
Розділення компонентів відбувається під час проходження крізь колонку парів проби разом із газом-носієм. Компоненти розподіляються між рухомим газом-носієм і нерухомою фазою, і переміщуються всередині колонки з різними швидкостями, які залежать від природи компонентів, що розділяють, природи нерухомої фази і температури. Після цього окремі компоненти суміші надходять, у порядку їх розташування в колонці, на детектор. Сигнал детектора залежить від концентрації компонента. Для автоматичного запису сигналу, детектор підключають до реєстру вального приладу (комп’ютер).
Рідинно-високошвидкісна хроматографія (РВХ) використовують для розділення розчинних нелетких сполук. Рухома фаза подається в колонку під тиском від 2 до 40 МПа і більше. Колонки виготовляють із нержавіючої сталі діаметром 1 – 3мм, довжиною 0,5 – 1,0м. Зразок вводять мікрошприцем. Найчастіше використовують УФ-детектор із λ = 254нм (при цій довжині хвилі поглинаються всі ароматичні та органічні речовини).
Використовують також детектор, який визначає кут заломлення світла, порівнюють значення кута заломлення світла чистого розчинника із значенням кута заломлення суміші, яка вийшла з колонки.
Позитивне: РВХ дає велику швидкість розділення і низьку концентрацію визначення (10-9г).
|
Мал. 4.3
|
|
|||||
|
|
|
|||||
|
Мал. 4.4
|
|
|||||
Питання |
Газова хроматографія |
Газорідинна хроматографія |
Рідинна хроматографія |
||||
Рухома фаза |
газ (N2, He, H2) |
газ (N2, He, H2, Ar) |
рідина (досліджувана суміш) |
||||
Нерухома фаза |
|
рідина з високою температурою кипіння (алкан з довгим ланцюгом), що підтримується твердим носієм (порувата інертна речовина – глинозем, SiO2, вугілля). |
тверда (дрібнозернисті речовини-носії) |
||||
Ввід проби |
у вигляді пари |
вприскують рідину медичним шприцом |
розчин рідкий |
||||
Результат |
газова хроматограма |
газова хроматограма (послідовність піків) |
хроматограма на папері (на пластинці) |
||||
Приклади |
газотвердофазна газо адсорбційна газорозподільна |
газорідинна |
|
||||
Застосування |
ідентифікація та визначення органічних і неорганічних сполук |
кількісне визначення інсектицидів, що містять галогени, в різних продуктах, визначення S, P, N – в досліджуваних речовинах |
для швидкого виявлення різних домішок, забруднень і натуральних компонентів в харчових продуктах |
||||
Примітка |
|
|
просування суміші через поруватий носій відбувається під:
|
||||
Колоночна хроматографія |
Площинна хроматографія |
|
|||||
Нерухома фаза: твердий адсорбент (Al2O3, глинозем, іонообмінна смола) Рухома фаза: розчинник (розчин електроліту), досліджуваний розчин. Процес пропускання розчинника крізь колонку – елюірування. Розчинник – елюент, видаляють перегонкою. Розділення суміші на окремі компоненти відбувається по мірі поступового переміщення вздовж колонки (мал.4.5; 4.6). Якщо компоненти суміші забарвлені, то їх розділення можна спостерігати візуально (внутрішня хроматографія).
Використовують іоніти (речовини, що здатні до обміну іонами). Іоніти складаються з матриці (R) та активних груп з рухомими іонами. Катіоніти – кислотні групи (сульфогрупи, карбоксильні), аніоніти – основні групи (аміногрупи, іміногрупи). При введенні в смолу групи, що селективно обмінюється з іонами розчину, одержують модифіковані іоніти. Наприклад, смола, що містить поліоксимні групи =N‑OH, є селективною стосовно до іонів Ni2+. Застосовують для:
Якщо білок ‑ позитивно заряджений, то він зв’язується на колонці з катіонообмінником (містить фенольні, сульфогрупи та карбоксильні групи). Якщо білок – негативно заряджений, то він зв’язується на колонці з аніонообмінником (представлений амінами або органічними основами). Позитивно заряджений білок знімається з колонки розчином NaCl або зміною рН буферу, яким проводять елювання. Білки з меншим позитивним зарядом вимиваються швидше, ніж білки з більшим позитивним зрядом.
|
Нерухома фаза: вода адсорбована на твердому носії (силікагель, целюлоза, крохмаль). Рухома фаза: органічний розчинник (або суміш розчинників).
Етапи проведення площинної хроматографії:
|
|
|||||
Rf
=
Хроматограма на папері
вихідні плями Х А В С
Пляма Х – досліджуваний зразок А, В, С – окремі компоненти Х складається з компонентів А і В. Розчинники підбирають залежно від природи речовин, носія та його полярності: носій – гідрофільна речовина; нерухомий розчинник – вода; рухомий розчинник – хлороформ, бутанол. Носії повинні бути нейтральними, нерозчинними у використовуваних розчинниках, повинні утримувати нерухому рідку фазу. Розподільну хроматографію на папері використовують для аналізу пептидів і амінокислот. Адсорбент – ниті целюлози (тверда нерухома фаза), розчинник – суміш: бутиловий спирт - оцтова кислота – вода. Хроматограму проявляють, висушують, аналізують знаходження досліджуваних компонентів. За інтенсивністю забарвлення та площею забарвлення плям роблять кількісний і якісний аналіз речовин. |
|
||||||







 фронт
розчин фронт розчинника
фронт
розчин фронт розчинника
 кінцеві
плями
кінцеві
плями
Мал.4.5 |
Мал.4.6 |
|
|

У катіонообмінній смолі відбувається реакція:
R‑COOH + K+ R‑COOK + H+
В аніонітах реакція іонного обміну відбувається за схемою:
R‑NH3OH + An- R‑NH3 An +OH-
Катіони легко елюються розбавленим розчином НСІ, амінокислоти сорбуються аніонітами і вимиваються розчином аміаку. Промивання водою завершає регенерацію.
Останні роки іоніти використовуються у вигляді плівок (мембран).
Прийоми розділення речовин в колонках


Внутрішня хроматографія (сорбент із зонами). Пробу пропускають через колонку – компоненти суміші розподіляють вздовж сорбенту, утворюючи зони. За забарвленням зон встановлюють якісний склад суміші. Приклад: розділення хлорофілу на пігменти.
речовина С
речовина А
|
Фронтальна хроматографія Колонку промивають розчинником, пропускають розчин суміші речовин (елюат). 1 речовина – з найменшою здатністю до сорбції Потім суміш: 1 речовини + 2 речовини, 1 речовина + 2 речовина + 3 речовина Можна виділити у чистому вигляді лише один компонент Застосовують для концентрування домішок, для виділення тільки одного компоненту суміші.
С
А+В
А
t(V) |
Елюентна хроматографія (найбільш поширена) В колонку вводять порцію суміші, промивають розчинником (елюентом). Речовини рухаються разом з розчинником з різними швидкостями згідно їх адсорбційної здатності. Результат: речовини з’являються в порядку зростання їх сорбційних властивостей. 1 речовина – найменша сорбційна здатність Остання речовина – найбільша сорбційна здатність. С
t(V) |
|
|
|




 речовина
В
речовина
В


 А+В+С
А+В+С

 А
А В
В
Лекція № 21. Рефрактометричний метод аналізу
Основні питання:
Характеристика рефрактометричного методу аналізу.
Показник заломлення. Спосіб вимірювання показника заломлення методом граничного кута.
Застосування рефрактометричного методу аналізу.
Промінь світла, що переходить з одного середовища до іншого, частково відбивається від поверхні розділу, а частково переходить в інше середовище, змінюючи при цьому свій початковий напрям. Зміна напряму прямолінійного поширення світла при переході з одного середовища в інше - заломлення або рефракція.
|
Мал. 4.7. Заломлення світлового променя на межі розділу двох прозорих однорідних середовищ (А); повне внутрішнє відбивання (В) |

Заломлення
світла (оцінюється за величиною показника
заломлення, позначається n) дорівнює
відношенню синуса кута падіння α до
синуса кута заломлення β:
![]()
Метод аналізу, який ґрунтується на визначенні показника заломлення досліджуваного розчину, називається рефрактометричним.
Показник заломлення (n) залежить від:
довжини хвилі падаючого світла (λ, n); Для видимого світла найбільший показник заломлення відповідає фіолетового випромінюванню (λ = 390 – 420нм).
температури (Т, n);
тиску (для газів).
Лінія |
Довжина хвилі, в ммк |
Позначення показника заломлення |
Показник заломлення води при 200С |
Синя лінія водню (лінія F) |
λF = 486 |
nF |
1,3371 |
Жовта лінія натрію (лінія D) |
λD = 589 |
nD |
1,3330 |
Червона лінія водню (лінія С) |
λC = 656 |
nC |
1,3311 |
Позначаючи показник заломлення, вказують довжину хвилі падаючого світла і температуру при якій проводиться вимірювання.
Запис nD20 означає, що показник заломлення був виміряний при 200С, довжина хвилі падаючого світла 589 ммк.
Абсолютний показник заломлення дорівнює добутку виміряного значення показника заломлення на абсолютний показник заломлення повітря: n = 1,00027 при тиску, рівному 101325 Па і кімнатній температурі 20 С.
N = 1,00027∙n
Показник заломлення залежить від ступеня поляризації молекули, її дипольного моменту, тобто абсолютний показник заломлення відображає особливості будови молекули досліджуваної речовини і пов’язаний з діелектричною проникністю середовища ε рівнянням: ε = n2.
Для виключення впливу зовнішніх факторів на значення показника заломлення, вводять поняття мольної і питомої рефракції. Мольна рефракція обчислюється для 1 молю, а питома рефракція – для 1 граму речовини.
Мольну рефракцію застосовують для:
Обчислюють за формулою:
R – мольна рефракція, см3/моль; - густина речовини; Мr – відносна молекулярна маса. |
Питому рефракцію застосовують для:
Обчислюють за формулою:
r – питома рефракція, см3/г; n – показник заломлення. |
В аналізі частіше використовують показник заломлення, який можна визначити за допомогою рефрактометра.
Метод визначення показника заломлення світла за значенням граничного кута заломлення або повного внутрішнього відображення використовується в приладі – рефрактометрі.
Рефрактометр

З боку передньої кришки корпусу розміщена шкала рефрактометра.
На вісі приладу закріплені:
рукоятка з окуляром для спостереження межі світлотіні та поєднання її з перехрестям сітки;
лімб дисперсії для запобігання забарвленості границі світлотіні;
механізм наведення, що міститься в корпусі.
|
|
Мал.4.8. Рефрактометр УРЛ: а – будова приладу (1 – корпус; 2 – камера, що складається з двох половин: нижньої нерухомої, верхньої рухомої; 3 – освітлювач; 4 – окуляр; 5 – шкала; 6 – дисперсний компенсатор; 7 – ручка; 8 – штуцер для термометра); б – показники внутрішньої шкали (при спостереженні в окуляр) |

Оптична схема рефрактометра
Досліджуваний розчин вміщують між площинами двох призм – освітлювальної та вимірювальної. Від джерела світла конденсором промінь світла спрямовується на вхідну площину освітлювальної призми, потім він проходить через тонкий шар досліджуваної речовини та заломлюється на межі досліджуваної речовини та площини вимірювальної призми. Промені, що вийшли з вимірювальної призми, фокусуються об’єктивом зорової труби в її полі зору, утворюючи світлу та темну частини поля, що розділені прямою лінією. Межею світлотіні є граничні промені. Для фіксації положення межі світлотіні відносно нерухомої шкали зорова труба повертається відносно вісі. Через окуляр зорової труби спостерігають: лінія світлотіні, перетин сітки та шкала. Для компенсації дисперсії променів, які вийшли з вимірювальної призми в зоровій трубці встановлені дві призми, що повертаються відносно вісі зорової трубки. Шляхом обертання призми встановлюють в таке положення, при якому межа світлотіні не має спектрального забарвлення. Відлік за шкалою здійснюється після усування спектрального забарвлення межі світлотіні в положенні перетину межі світлотіні і сітки.











Лекція № 22. Люмінесцентний аналіз
Основні питання:
Походження люмінесценції.
Характеристики та закономірності люмінесценції.
Апаратура.
Застосування люмінесценції.
Люмінесценція – світіння атомів, молекул та інших частинок, що виникає в результаті електронного переходу при повертанні із збудженого стану до основного.
Залежно від способу збудження частинок розрізняють наступні види люмінесценції:
Спосіб збудження |
Вид люмінесценції |
Електромагнітне випромінювання (УФ, видиме світло) |
фотолюмінесценція |
Енергія хімічних реакцій, що відбуваються в живих організмах |
біолюмінесценція |
Рентгенівське випромінювання |
рентгенолюмінесценція |
Енергія хімічних реакцій |
хемілюмінесценція |
Електрична енергія |
електролюмінесценція |
В аналітичній хімії найчастіше використовують фотолюмінесценцію.
Молекула, поглинаючи квант світла, переходить з основного стану S0 у збуджений електронний стан S1. За кімнатної температури молекули знаходяться в основному коливальному стані. При переході у збуджений стан молекула потрапляє на один з його коливальних рівнів. Поглинання молекулою кванта світла відбувається протягом 10-15 сек. Потім за 10-12 сек відбувається перехід електрону на нижній коливальний підрівень збудженого стану (коливальна релаксація).
Повернення молекули з нижнього коливального стану S1 до не збудженого стану S0 може відбутися 3 шляхами:
втрата молекулою енергії у вигляді теплоти при зіткненні з другими частинками (внутрішня конверсія);
повертання молекули на будь-який коливальний підрівень основного стану з випромінюванням енергії у вигляді кванта світла без зміни спіну електрону (флуоресценція);
перехід молекули із збудженого стану S1 у метастабільний стан Т1, а потім до основного стану S0 (або в результаті внутрішньої конверсії з виділенням теплоти; з виділенням кванта світла – фосфоресценція).
Синглетний стан – стан із сумарним спіном електрона, що дорівнює нулю (електрони, що знаходяться на одній орбіталі антипаралельні).
Триплетний стан – стан, в якому спіни збудженого електрона та електрона, що знаходиться в основному стані – паралельні. Сумарний спін дорівнює 1.
Електронні переходи :
синглет – синглетні (баз зміни спіну електрона), відбуваються при поглинанні світла, при флуоресценції;
синглет – триплетні (із зміною спіну електрону заборонені згідно правила відбору). Такі переходи (інтеркомбінаційна конверсія) відбуватимуться за спеціальних умов (у присутності важких атомів). Термін життя триплетного стану – 10-3 – 10-2 сек. Молекули в триплетному стані легко втрачають свою енергію у безвипромінювальних процесах – дезактивуються з виділенням теплоти. Дезактивація може відбутися при взаємодії молекули в триплетному стані з молекулами, що мають неспарені електрони (молекули кисню), а також при зіткненнях з іншими частинками.
Термін світіння є основною характеристикою люмінесценції.
За терміном світіння розрізняють:
флуоресценцію (коли світіння припиняється разом із припиненням збудження);
фосфоресценцію (коли світіння триває протягом деякого часу після припинення збудження).
Висновок:
Флуоресценція спостерігається частіше, ніж фосфоресценція. Інтенсивна фосфоресценція спостерігається в органічних сполуках, що знаходяться в замороженому або у скловидному станах (дифузія мінімальна).
Переваги люмінесцентного аналізу:
найбільш чутливий метод (використовують для визначення слідів елементів), дозволяє визначати 10-10-4 мкг∙мл-1 речовини;
вимірюють сам сигнал, і границя виявлення залежить від інтенсивності джерела і чутливості детектора (в спектрофотометрії вимірюють різницю 2-х сигналів І0 та Іl);
висока селективність (підвищують, змінюючи довжину хвилі збудження і реєстрації сигналу, час спостереження у фосфоресцентних методах, рН розчину, температуру тощо).
Закономірності люмінесценції

Для вимірювання флуоресценції використовують спектрофлуориметри і флуориметри, для вимірювання фосфоресценції – фосфориметри.
Основні вузли:
Джерела збудження (ртутно-кварцеві лампи, ксенонові лампи, вольфрамогалогенідні лампи, які дають випромінювання в УФ- та видимій областях спектру);
Прилади для виділення спектрального діапазону:
1 прилад застосовують для виділення смуги випромінювання збуджуючої речовини.
2 прилад використовують для виділення потрібної довжини хвилі (або інтервалу довжин хвиль) з спектру люмінесценції.
Використовують призменні та дифракційні монохроматори (спектрофлуориметр) та світлофільтри (флуориметр).
Детектори (використовують фотопомножувачі, що перетворюють світловий сигнал в електричний, та лічильники фотонів.
Світло від лампи → первинний світлофільтр (пропускає випромінювання з довжинами хвиль збудження) → кювета з досліджуваним розчином (люмінесцентне випромінювання) → другий світлофільтр (пропускає люмінесцентне випромінювання та затримує розсіяне та збуджуюче випромінювання) У приладі джерело збудження, кювету з досліджуваним розчином і детектор розміщують залежно від способу вимірювання випромінювання:
|
|

Застосування люмінесценції
Для
визначення органічних речовин (білки,
вітаміни, наркотики, ліки) (для
визначення РЗЕ, домішок у напівпровідникових
матеріалах)В неорганічному аналізі



(метод
базується на здатності окремих речовин
випромінювати люмінесценцію при
освітленні їх ультрафіолетовими
променями за умови видалення видимого
світла) Визначення
ступеня свіжості м’яса;
На
початку псування м’ясо змінює
люмінесценцію: на загальному тлі
світіння з’являються точки, що світяться
специфічно. Несвіже м’ясо має тьмяний,
темно-червоний, нерівномірний, з багатьма
світними точками і зеленими плямами
колір люмінесценції. Якщо для аналізу
беруть м’ясний екстракт яловичини: свіже
м’ясо – темно-зелений
колір
люмінесценції; з
початковими ознаками псування –
зелено-блакитний;
несвіже
м’ясо – блакитний
колір
люмінесценції.
Визначення
видової приналежності м’яса та
м’ясопродуктів;
Вид
м’яса (субпродукту)
Колір
люмінесценції
Вид
м’яса (субпродукту)
Колір
люмінесценції
Яловичина
Темно-червоний,
червоно-фіолетовий
Жирові
тканини
Світло-жовтий
Баранина
Темно-коричневий
Кістки,
сухожилля, хрящі
Світло-блакитний
Свинина
Рожевий
з коричневим відтінком
Серце
Злегка
блакитнуватий
Телятина
Світло-коричневий
Печінка
Яскраво
салатовий
Легені
Голубувате
- волошковий
Нирки
Зеленкуватий За
кольором люмінесценції встановлюють
сорт борошна: чим більше в ній висівок,
тим інтенсивніше світіння.В контролі якості продукції харчування
Лекція № 23. Електрохімічні методи аналізу
Основні питання:
Електрохімічні методи аналізу, їх класифікація.
Сутність потенціометрії.
Потенціометричне визначення рН розчинів.
В основі електрохімічних методів аналізу і досліджень є процеси, які відбуваються на електродах або в між електродному просторі.
Для аналізу використовують залежність:
Сили струму І, мА ‑ вольтамперометрія;
Потенціалу Е, В – потенціометрія;
Потенціометричний аналіз ґрунтується на вимірюванні потенціалів електродів залежно від концентрації іонів, що визначаються, у відповідній окислювально-відновній системі.
Потенціометрія об’єднує групу методів визначення різних фізико-хімічних величин і кількісного аналізу розчинів на основі вимірювання ЕРС (електрорушійної сили).
Електролітичної провідності œ (питома електропровідність), Ом/см – кондуктометрія;
Опору R
від концентрації аналізованого розчину, або вимірюють ці параметри з метою встановлення кінцевої точки титрування речовин, яку визначають.
Електрохімічні методи аналізу базуються на застосуванні:
Законів Фарадея (1833р.), які встановлюють кількісний зв’язок між хімічними та електричними величинами: кількість речовини, що виділилася або розклалася на електродах, прямо пропорційна кількості електрики, що пройшла через розчин.
Закону Ома;
Рівняння електродного потенціалу Нернста (1888р.), що зв’язує рівноважний електродний потенціал з концентраціями реагуючих речовин.
![]() ,
де
,
де
φ0 – стандартний електродний потенціал;
R – універсальна газова стала, дорівнює 8,31 Дж/(моль∙К);
Т – абсолютна температура;
F – стала Фарадея, дорівнює 9,65∙104 Кл/моль;
Величина електродного потенціалу залежить від:
температури;
природи металу і природи розчинника;
активності іонів у розчині (молярної концентрації);
За температури 250С (298К) і відповідних значеннях R, F і коефіцієнту переходу від натуральних логарифмів до десяткових рівняння Нернста має вигляд:
![]()
Стандартний електродний потенціал визначається як ЕРС елемента, складеного з досліджуваного електрода і нормального водневого електрода за умови, що концентрації (активності компонентів) потенціал визначальної реакції, яка відбувається на визначальному електроді, дорівнюють одиниці.
У потенціометрії широко використовується вимірювання потенціалу досліджуваного електрода відносно порівняльного електрода. Потенціали виражаються в шкалах або вибраного електрода, або перераховуються на водневу шкалу, яка відповідає стандартному (нормальному) водневому електроду порівняння [(Pt) H2 │ 2H+ (p = 1атм = 101,325 кПа), Н+ (а = 1)].
Чим метал активніший, тим більш від’ємний рівноважний потенціал він буде мати. Рівноважне значення електродного потенціалу визначається рівнянням Нернста.
Електроди І роду ‑ активні металеві електроди (Аg, Cu, Cd, Pb), потенціал яких залежить від активності власних іонів у розчині. Належить скляний електрод.
Активні металеві електроди можна використовувати для визначення не тільки власних іонів металу, але й для визначення аніонів, які утворюють із цими металами слабкорозчиннні солі.
Електроди ІІ роду – електроди, потенціал яких визначається концентрацією аніонів. Вони представляють з себе метал, вкритий шаром малорозчинної солі цього металу (хлорсрібний електрод).
Електрохімічні методи аналізу


(базується
на вимірюванні кількості продукту
реакції, реактив – електрони, які
підводяться до поверхні електроду) Електролітичне
осадження металів
(виділення металів електричним струмом
і зважування одержаного на електроді
осаду металу); Внутрішній
електроліз
(для визначення незначних домішок
сторонніх металів) – не застосовується
зовнішнє джерело струму; Розділення
металів електролізом із застосуванням
ртутного катода (осадження
полегшується утворенням
амальгам). (без
хімічної реакції) Потенціометрія
(в досліджуваний розчин занурюють
водневий електрод і за показниками
потенціометра визначають концентрацію
водневих іонів (рН розчину) – вимірюють
потенціал; Кондуктометрія
(безпосереднє вимірювання електропровідності
розчину), судять про вміст
електроліту,
води в концентрованих кислотах.
Широко
застосовуються для автоматизації
контролю виробництва.
Електрогравіметрія (електроваговий аналіз)
Безпосереднє вимірювання електрохімічних властивостей досліджуваної речовини

(при
титруванні спостерігають за зміною
електрохімічних властивостей розчину) Потенціометричне
титрування
(метод визначення точки еквівалентності
за зміною окислювального потенціалу
індикаторного електрода);
Застосовують
для визначення: загальної
концентрації розчину, кількох речовин
в одному розчині без їх попереднього
розділення, проводять титрування
каламутних і забарвлених середовищ. Кондуктометричне
титрування
(метод визначення точки еквівалентності
за зміною електропровідності розчину);
Застосовують
для визначення:
розчинності та ДР важкорозчинних
сполук; іонного добутку води; концентрації
забарвлених і каламутних розчинів; при
аналізі суміші електролітів, для
швидкого визначення клінічного стану
організму. Амперометричне
титрування
(зміна сили полярографічної сили
струму);
Застосовують
для визначення:
концентрації вітамінів, гормонів,
амінокислот у препараті; для дослідження
процесу обміну кисню при фотосинтезі
та диханні; як додатковий метод при
діагностиці ракових захворювань.
Об’ємні електрохімічні методи
Потенціометричне
титрування (мал.4.10) (базується
на вимірюванні потенціалу індикаторного
електрода, зануреного в досліджуваний
розчин. Точку еквівалентності визначають
за зміною електродного потенціалу в
кінцевій точці титрування. Застосовують
прилади – потенціометри.)
Індикаторний
електрод
– електрод вимірювання, потенціал його
залежить від концентрації досліджуваних
іонів у розчині. Належать: водневий,
скляний, хінгідронний, сурм’яний,
платиновий, срібний, ртутний електроди
(мал..4.9).
Електрод
порівняння
(стандартний електрод) – відносно якого
вимірюють потенціал індикаторного
електрода. Потенціал електрода порівняння
залишається сталим при зміні концентрації
досліджуваних іонів.
Переваги: значна
точність; добра
відтворюваність; різка
зміна потенціалу індикаторного
електрода поблизу точки еквівалентності; відсутність
індикаторної погрішності (відсутність
органічного індикатора).



Іонометрія

рН–метрія
(потенціометричне визначення рН)
Складають
гальванічний елемент з 2-х електродів:
стандартний (електрод порівняння) –
φ=const; електрод визначення (індикаторний
електрод) – φ залежить від С (активності)
іонів водню у розчині.
Індикаторний
електрод
– скляний електрод (найчастіше), добре
працює у будь-яких агресивних середовищах,
і будь-якому діапазоні рН, не чутливий
до окислювально-відновних процесів,
індиферентний до ПАР, білків).
Прилади:
потенціометри, шкала яких проградуйована
в одиницях рН (відпадає потреба у
побудові калібрувального графіка);
рН-340, рН-262, рН-673, П-261, іономір ЕВ-74.
Переваги: більш
точний, ніж індикаторний (дає змогу
виміряти рН з точністю 0,02-0,05); неруйнівний
(для вимірювання рН тканин і клітин,
контроль кислотності безпосередньо
у травній системі людини, контроль
газів в крові); дає
можливість вимірювати рН багатокомпонентних
систем і забарвлених розчинів.
Мал.4.9. Будова хлор срібного та напівскляного електродів |
Мал.4.10. Потенціометричне титрування |
|
|
|
|

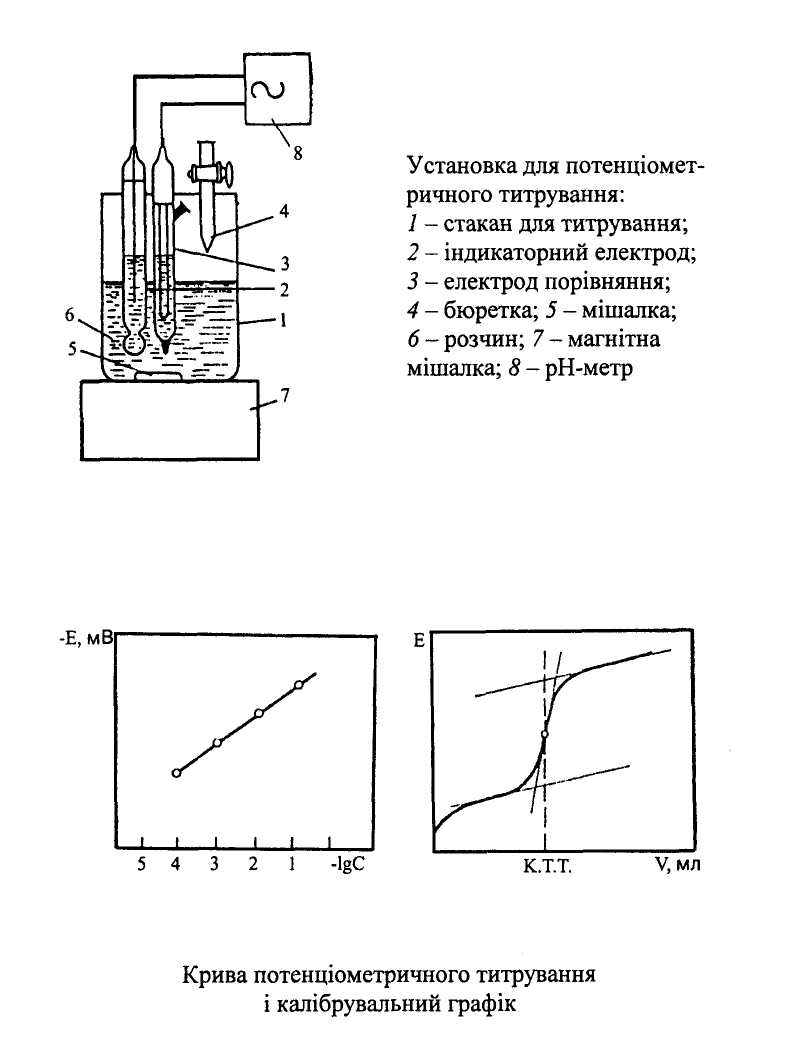
Висновки:
Для аналітичних потенціометричних визначень в досліджуваний розчин потрібно занурити два електроди: робочий (індикаторний) електрод – за потенціалом якого визначають концентрацію іонів у розчині та електрод порівняння (стандартний) електрод – відносно якого вимірюють потенціал індикаторного електрода.
Пряма потенціометрія ґрунтується на прямому визначенні потенціалу індикаторного електрода і обчисленню концентрації визначуваного іона за рівнянням Нернста. Цей метод широко використовується в рН-метрії. Найчастіше використовують скляний електрод.
Скляний електрод – іонно-селективний електрод мембранного типу. Воднева функція скляного електрода пояснюється особливим хімічним складом скла. Інтервал вимірювання рН = 0,5 – 12. Вимірювання рН застосовують в технохімічному контролі харчових продуктів в їх виробництві, де біохімічні та мікробіологічні процеси дуже залежать від рН середовища.
У потенціометрії поширені іонно-селективні електроди – це індикаторні електроди, які мають високу специфічність до певних іонів (електроди з твердими іонними мембранами, рідкі іонні мембрани, газові електроди).
Іонно-селективна потенціометрія використовується для аналізу харчових продуктів: NaCІ, нітрат-іонів, іонів кальцію – у дитячому харчуванні, фторид-іонів – у питній воді; йодид-іонів‑ в морській капусті, хлорид-іонів – у молоці, сирах, маргарині, хлібі.
Потенціометричне титрування – це варіант титриметричного аналізу, в якому кінцеву точку титрування визначають за різкою зміною потенціалу індикаторного електрода поблизу точки еквівалентності.
Технічна характеристика рН-метра рН-340
Прилад застосовують для визначення рН розчинів, окислювально-відновних потенціалів, а також застосовують при проведенні потенціометричного титрування та як високоомний мілівольтметр.
Границі вимірювання рН на приладі від -1 до 14. Температура навколишнього середовища від 10 до 350С.
рН-метр рН-340 – настільний прилад в комплекті з датчиком ДЛ-02 і електродами вимірювання та порівняння. Всі органи управління приладу виведені на зовнішню панель, шкала приладу цифрована в одиницях рН. Як вимірювальні електроди використовують скляні електроди типу ЕСЛ-41Г-04; ЕСЛ-41Г-05; ЕСЛ-11Г-04; ЄСЛ-11Г-05 та інші, які передбачені для роботи в комплекті з хлорсрібним електродом порівняння. Електроди і термометр закріплюються зажимами до утримувача вертикального штативу з підставкою, на яку встановлюється стакан з досліджуваним або буферним розчином.
Перед проведенням вимірювання рН розчинів скляний електрод вимочують не менше 8 год. в стакані з розчином НСІ, С (НСІ) = 0,1 моль/л.
Перед зануренням електродів у досліджуваний розчин необхідно промити їх дистильованою водою, надлишок води видалити з поверхні електродів фільтрувальним папером.
Після закінчення роботи з приладом електроди повинні залишатися зануреними у воду або 0,1Н розчин НСІ. Не можна допускати висихання скляного електрода, перебування тривалий час у розчині кислоти (зміна характеристики електрода).
У випадку утворення плівок на електродах їх можна видалити промиванням електродів органічними розчинниками, кислотами або лугами. Після промивання електродів цими розчинами їх необхідно ретельно промити водою, а потім показники приладу необхідно перевірити за буферними розчинами, які готуються з фіксаналів кваліфікації „для рН-метрії”. Настройка приладу проводиться за двома стандартними розчинами 1,68 рН та 9,22 рН (фіксанали цих буферних розчинів йдуть в комплекті з приладом). При використанні приладу слід мати на увазі, що буферні розчини при багаторазовому використанні можуть змінювати значення рН, тому їх необхідно періодично заміняти свіжевиготовленими.
Відлік показників у широкому діапазоні проводять за нижньою шкалою приладу, в будь-якому вузькому діапазоні рН відлік показників проводять за верхньою шкалою при відповідному положенні перемикача „границя виміру”. Відлік проводять так: значення рН, що вимірюють дорівнює сумі початкового показника рН для цього діапазону і показника верхньої шкали приладу.
Лекція № 24. Метрологічні основи аналітичної хімії. Статистична обробка результатів аналізу
Основні питання:
Основні метрологічні характеристики.
Спосіб зовнішніх стандартів.
Погрішності та невизначеність вимірювань. Точність і її складові.
Класифікація помилок.
Визначення грубих промахів. Q-тест
Статистична обробка результатів аналізу.
Метрологія – наука про методи вимірювання величин.
Основні метрологічні характеристики
Будь-який експериментатор при проведенні вимірювань одержує серію даних, що спричинено різними факторами.
Вивченням загальних питань, пов’язаних з вимірюванням, обробкою та інтерпретацією результатів хімічного аналізу займається спеціальний розділ аналітичної хімії ‑ хімічна метрологія.
Вимірювання – це визначення числового значення вимірювальної величини, вираженого в певних одиницях.
Правильність вимірювання – це якість вимірювання, яка відображає наближеність до нуля систематичної погрішності, визначає ступінь наближення результату аналізу до дійсного вмісту речовини.
Відтворюваність вимірювань – якість вимірювань, яка відображає близькість один до одного результатів вимірювань, що виконуються за різних умов, характеризує відхилення окремих вимірювань від їх середнього арифметичного значення.
Точність вимірювань – це якість вимірювань, яка відображає близькість їх результатів до справжнього значення вимірюваної величини. Кількісно точність виражається оберненою величиною модуля відносної погрішності. Якщо, відносна погрішність вимірювання характеризується значенням 0,01%, то точність дорівнюватиме 1/10-4 = 104.
За допомогою приладів можна проводити прямі та непрямі вимірювання.
При прямому вимірюванні експериментатор безпосередньо одержує числове значення вимірюваної величини. Наприклад, при відліку за шкалою бюретки визначають об’єм розчину в мілілітрах; за шкалою фотоелектроколориметра знаходять значення оптичної густини досліджуваного розчину. Виміряна величина може включати випадкову помилку.
Непрямим вимірюванням називають вимірювання, на основі якого визначають нову величину за певною формулою. Обчислена величина – функція безпосередньо виміряної величини (декількох вимірюваних величин). Кожна з цих величин має свою помилку. Ці помилки по різному впливають на помилку результату аналізу.
Існує багато фізичних величин, доступних прямим вимірюванням і функціонально пов’язаних із вмістом речовини.
m = M·n (маса будь-якої чистої речовини пропорційна її кількості, молярна маса – коефіцієнт пропорційності), використовується в гравіметрії.
![]() (при
титруванні кількість досліджуваної
речовини пов’язано з об’ємом стандартного
розчину титранту).
(при
титруванні кількість досліджуваної
речовини пов’язано з об’ємом стандартного
розчину титранту).
A = ε·l·c (в забарвлених розчинах існує взаємозв’язок між концентрацією речовини, що поглинає світло, та оптичною густиною), використовується в фотоелектроколориметрії та методах візуальної колориметрії.
Будь-яка механічна, оптична, електрична величина, що може бути пов’язаною з вмістом речовини, і може бути використана для її визначення, називається аналітичний сигнал.
Функціональний зв’язок між аналітичним сигналом і вмістом (наприклад, концентрацією) можна представити як
y = f(c)
Функція f, що пов’язує вміст і аналітичний сигнал, називається градуювальною функцією.
Загальна схема вимірювання вмісту речовини складається з:
Установлення градуювальної функції f.
Вимірювання аналітичного сигналу досліджуваного зразка y.
Знаходження за величиною y за допомогою функції f вмісту досліджуваного компонента c.
Таким чином, всі вимірювання хімічних величин є непрямими і базуються на використанні градуювальної функції.
Для проведення хімічного аналізу необхідно знати точний вигляд градуювальної функції.
Для окремих методів аналізу точний вид градуювальної функції відомий з теорії: в гравіметричному аналізі використовується залежність m = M·n (параметр – молярна маса, встановлений з високою точністю). Подібні методи не потребують експериментального визначення калібрувальної функції, називають абсолютними. Більшість методів належить до відносних, градуювальна функція встановлюється експериментально, як правило, перед проведенням аналізу, так як може сильно залежати від умов проведення аналізу.
Процедуру експериментальної побудови градуювального (калібрувального) графіка називають градуюванням (калібруванням)
Таким чином, абсолютні методи аналізу не потребують побудови градуювального графіка, а у відносних методах – обов’язкова.
Для виконання градуювання необхідно мати набір зразків з надійно встановленим вмістом досліджуваного компонента. В загальному випадку такі зразки називають зразками порівняння (ЗП). До групи зразків порівняння належать стандартні зразки (СЗ).
Стандартні зразки – це спеціально виготовлений матеріал, склад якого надійно встановлений і юридично забезпечений: кожний СЗ має офіційний документ (паспорт, атестат), виданий уповноваженим органом системи Держстандарту в якому містяться дані про його склад (як правило, про вміст всіх макрокомпонентів і найважливіших мікрокомпонентів). Наприклад, при атестації нової методики використання СЗ є обов’язковим.
Величини аналітичних сигналів та конкретний вид градуювальної функції залежать від умов, за яких проводиться вимірювання.
Найважливішою вимогою до процесу градуювання є забезпечення максимально точного дотримання однакових умов градуювання та проведення дослідження зразка: градуювання і аналіз необхідно виконувати на тому ж самому приладі, за однакових значеннях інструментальних параметрів, також інтервал в часі між градуюванням і аналізом повинен бути коротким.
Крім того, якщо на величини аналітичних сигналів впливають сторонні компоненти зразка (його матриця) або його фізичний стан, то ЗС, що використані для градуювання, повинні як найбільше відповідати досліджуваному зразку з точки зору цих параметрів.
Тому СЗ, дуже часто імітують типові об’єкти аналізу (наприклад, СЗ ґрунту, харчових продуктів, природних вод, рудних концентратів). Використовують також спеціальні прийоми градуювання, що забезпечують максимальну адекватність її умовам аналізу.
Спосіб зовнішніх стандартів
Найбільш простий і поширений спосіб градуювання – спосіб зовнішніх стандартів, або спосіб «градуювального графіка». В цьому способі беруть ряд СЗ із вмістом досліджуваного компонента c1, c2, ... cn, проводять з ними всі необхідні згідно з методикою аналітичні операції та вимірюють їх аналітичні сигнали (y1, y2, ... yn, відповідно). За одержаними параметрами експериментальних значень (ci, yi) будують залежність y від c и апроксимують її підходящою алгебраїчною функцією або графічно. При цьому вибирають такі умови аналізу, об ця залежність була б лінійною. Потім аналізують невідомий зразок, вимірюють його аналітичний сигнал yx. З використанням одержаної градуювальної функції знаходять (алгебраїчно або графічно) відповідне значення cx. Наприклад, у випадку лінійної градуювальної функції, що описана рівнянням:
y = kc + b, невідомий вміст можна знайти. Величина b є значенням аналітичного сигналу при нульовій концентрації визначуваного компонента, називається фоновим значенням сигналу. Вона важлива при оцінюванні чутливості методик.
Іноді спосіб зовнішніх стандартів додатково спрощують, скорочуючи число СЗ до двох (спосіб обмежуючих розчинів), або навіть одного (спосіб одного стандарту).
У способі обмежуючих розчинів лінійний (в обраному концентраційному діапазоні) характер градуювальної функції постулюють заздалегідь (та, за можливості, експериментально перевіряють), а СЗ обирають таким чином, щоб c1<cx<c2.
![]()
Якщо c1 та c2 достатньо близькі до cx, то спосіб обмежуючих розчинів іноді дає більш точні результати, ніж "повний" варіант способу зовнішніх стандартів.
У способі одного стандарту припускають прямо пропорційний вигляд градуювальної функції y = kx (без вільного члена, фоновий сигнал відсутній). В цьому випадку:
![]()
В будь-якому варіанті способу зовнішніх стандартів СЗ готують і використовують окремо від досліджуваного зразка. Тому склад і властивості СЗ не завжди достатньо точно відповідають істинним значенням для досліджуваної проби. В окремих випадках це може спричинити значні погрішності результатів. У подібних ситуаціях слід використовувати спеціальні способи градуювання.
Погрішності та невизначеність вимірювань. Точність і її складові
На будь-який процес вимірювання впливають різні фактори, які викривляють результати вимірювання. Різниця результату вимірювання від істинного значення вимірюваної величини називається погрішністю.
Так як будь-який результат вимірювання містить погрішність, точне значення вимірюваної величини ніколи не може бути установлено. Однак можливо вказати деякий діапазон значень, в межах якого може з певним ступенем достовірності знаходитися істинне значення. Цей діапазон називається невизначеність результату вимірювання. Оцінка невизначеності результатів хімічного аналізу є важливою задачею хімічної метрології.
В сумарну невизначеність результату вимірювання вносять вклад погрішності двох різних типів. Нехай в результаті однократного вимірювання деякої величини одержано значення x*, що відрізняється істинного значення x0. Повторимо вимірювання ще декілька разів.
Можливі варіанти взаємного розміщення серії виміряних значень та їх істинного значення показані на мал.1. У першому випадку (мал. 1, б) має місце зміщення всієї серії даних (та її середнього) відносно істинного значення. Відповідна складова невизначеності називається систематичною погрішністю. У другому випадку (мал. 1, в) спостерігається розброс даних відносно середнього значення з результатів вимірювання. Така складова невизначеності називається випадковою погрішністю. В реальному випадку завжди має місце систематична та випадкова складові. Походження систематичних і випадкових погрішностей пов’язано з різною природою факторів, що впливають на процес вимірювання.
Фактори постійного характеру або ті, що мало змінюються від вимірювання до вимірювання спричиняють систематичні погрішності, фактори, що швидко змінюються спричиняють появу випадкових погрішностей.
Правильністю називається якість результатів вимірювання (або вимірювальної процедури в цілому), що характеризує малість (близькість до нулю) систематичної погрішності.
Відтворюваність (прецизіонність) ‑ якість, що характеризує малість випадкової погрішності. Тобто, правильність результатів – це їх незміщення, а відтворюваність – це їх стабільність.
Результати точні за умови малості як систематичної, так і випадкової погрішностей.
Отже, правильність і відтворюваність – це дві складові точності, називаються характеристиками точності.
В хімічній метрології традиційно оцінюють точнісні характеристики окремо.
x0 х*

 +
a
+
a
 ●●│● +
б
●●│● +
б
● ●
│ ● + ● в
●
│ ● + ● в
Мал. 1. Ілюстрація понять систематична і випадкова погрішність. Точки і зірочки ‑ результати одиничних вимірювань, вертикальні відрізки – середні значення.
Відтворюваність характеризує ступінь розсіяння даних відносно середнього значення. Тому для оцінки відтворюваності необхідно попередньо обчислити середнє x з серії результатів повторних (паралельних) вимірювань x1, x2, ... xn.
Зверніть увагу, що в серії значень повинні бути відсутніми промахи ‑ окремі значення, які різко відрізняються від інших, як правило, одержані в умовах грубого порушення вимірювальної процедури (методики аналізу). Тому спочатку (ще до обчислення середнього) за допомогою спеціальних статистичних тестів.
Як міру розбросу даних відносно середнього найчастіше використовують дисперсію та похідні від неї величини ‑ (абсолютне) стандартне відхилення та відносне стандартне відхилення.
В хімічному аналізі для характеристики відтворюваності найчастіше використовують відносне стандартне відхилення За допомогою відносних стандартних відхилень можна порівнювати між собою відтворюваності як різних даних, так і різних методик і методів аналізу.
Серед всіх методів хімічного аналізу найкраща відтворюваність (тобто найменші sr) характерна для класичних хімічних методів аналізу – гравіметрії та титриметрії. За оптимальних умов типові величини sr для них складають n.10-3 (десяті частки процента). Серед інструментальних методів найбільш високу відтворюваність має кулонометрія особливо в прямому варіанті (до n.10-4).
Більшість інших інструментальних методів характеризуються величинами sr від 0,005 до 0,10. Методи з нижчою відтворюваністю належать до напівкількісних методів. Незважаючи на невисоку точність, вони часто мають інші переваги: простота, експресність, економічність (тест-методи). Ці методи використовують, наприклад, для швидкої оцінки стану навколишнього середовища.
Вклад випадкової погрішності в загальну невизначеність результату вимірювання можна оцінити за допомогою методів теорії ймовірності і математичної статистики.
Із-за наявності випадкової погрішності одна й та же величина x при кожному наступному вимірюванні набуває нове значення. Такі величини називають випадковими. Випадковими величинами є не тільки окремі результати вимірювань xi, але й середні x (а також дисперсії s2(x) і всі похідні від них величини). Тому x може служити лише приблизною оцінкою результату вимірювання. В той же час, використовуючи величини x і s2(x), можливо оцінити діапазон значень, в якому із заданою ймовірністю P може знаходитися результат.
Ця ймовірність P називається доверительною ймовірністю, а відповідний інтервал значень ‑ доверительним інтервалом.
Строгий розрахунок границь доверительного інтервалу випадкової величини можливий лише за умови підпорядкування величини закону розподілу. Закон розподілу випадкової величини ‑ одно з фундаментальних понять теорії ймовірностей.
При обчисленні доверительного інтервалу постає питання щодо вибору доверительної ймовірності P. При дуже малих значеннях P висновки стають недостатньо надійними. Значення близькі до 1 брати теж недоцільно, так як в цьому випадку доверительні інтервали будуть сильно широкими, малоінформативними. Для більшості хіміко-аналітичних задач оптимальним значенням P є 0,95. Величина доверительного інтервалу сама по собі дозволяє охарактеризувати лише випадкову складову невизначеності.
Визначення грубих промахів. Q-тест
В серії даних, які підлягають статистичній обробці повинні бути відсутніми промахи (грубі помилки). Тому перед початком проведення обробки результатів аналізу, починаючи з обчислення середнього значення) необхідно з’ясувати чи містить серія значень грубі помилки, і якщо так, то виключити їх з розгляду. Для виявлення промахів існує статистичний тест, що називається Q-тестом.
Алгоритм виконання q-тесту:
Серію даних упорядковують по зростанню: x1 ≤ x2 ≤ ... ≤ xn-1 ≤ xn.
В якості можливого промаху розглядають одно з значень x1 або xn ‑ те, яке якнайдалі відстоїть від сусіднього значення (тобто для якого більша різниця x2 ‑ x1 або, відповідно, xn ‑ xn-1). Позначимо цю різницю як W1.
Розмах всієї серії, тобто різницю між максимальним і мінімальним значенням xn-x1, позначимо W0.
Тестовою статистикою є відношення:
![]() ,
де
,
де
W1 – різниця між двома сусідніми значеннями;
W0 – розмах всієї серії;
ξ – тестова статистика, знаходиться в межах від 0 до 1. Чим далі відстоїть "підозріле" значення від основної маси даних, тем вища ймовірність того, що цей промах - і тим більшою буде величина ξ.
Критичною величиною служить табличне значення Q-коефіцієнта Q (P, n).
Якщо тестова статистика перевищує критичну величину (ξ>Q), відповідне значення вважають промахом та з серії даних виключають.
Після цього слід перевірити на наявність промахів і інші дані (з другим значенням Q), оскільки промах в серії може бути не один.
При використанні Q-тесту замість стандартної доверительної ймовірності, рівної 0,95, використовують значення P = 0,90. Найбільш достовірні результати одержують при n = 5 ‑ 7. Для серій більшого або меншого розміру Q-тест є недостатньо надійним.
Таблиця
Q-коефіцієнти для ймовірності P = 0.90 та різних значень n
n |
Q |
n |
Q |
3 |
0,94 |
7 |
0,51 |
4 |
0,76 |
8 |
0,47 |
5 |
0,64 |
9 |
0,44 |
6 |
0,56 |
10 |
0,41 |
Приклад №1
При спектрофотометричному аналізі розчину органічного барвника одержані значення оптичної густини, що дорівнює 0,376; 0,398; 0,371; 0,366; 0,372 та 0,379. Чи містить ця серія промахи? Чому дорівнює середнє значення оптичної густини?
Розв’язок:
Розміщуємо отримані результати в порядку зростання:
0,366; 0,371; 0,372; 0,376; 0,379; 0,398
Різниця 0,371 – 0,366 = 0,005;
Різниця 0,398 – 0,379 = 0,019, тому кандидатом у промахи є значення 0,398.
W1 = 0,019.
Розмах вибірки W0 = 0,398 – 0,366 = 0,032.
Тестова
статистика
![]()
Критична величина Q (P = 0,90; n = 6) дорівнює 0,56. Отже, ξ>Q, значення 0,398 ‑ промах, його необхідно виключити.
Перевіряємо серію значень, які залишилися:
0,371 – 0,366 = 0,005;
0,379 – 0,376 = 0,003, тому наступний кандидат у промахи ‑ 0.366.
Маємо: W1 = 0,005; W0 = 0,379 – 0,366 = 0,013;
ξ
=
![]()
Q(P = 0,90; n = 5) = 0,64; ξ<Q, значення 0,366 не є промахом.
0,366; 0,371; 0,372; 0,376; 0,379 – серія даних
Середнє значення оптичної густини складає 0,373:
![]()
Обробка серії даних разом з промахом була б в цьому випадку грубою помилкою та спричинила серйозні викривлення значень.
Статистичну обробку результатів аналізу, одержаних дослідним шляхом проводять згідно наступної схеми:
Обчислення середнього арифметичного.
Дані, що одержані з n кількості вимірювань, утворюють ряд величин х1, х2, х3….хn.
З цієї серії вимірювань знаходять границі відхилення.
![]() .
.
де хі – дані, що одержані при проведенні вимірювань;
![]() ‑ середнє
арифметичне, що обчислюється за формулою:
‑ середнє
арифметичне, що обчислюється за формулою:
![]() .
.
Сума
всіх позитивних і негативних відхилень
від середнього повинна дорівнювати
нулю:![]() .
Це виконується тим точніше, чим більш
точно обчислено значення
.
.
Це виконується тим точніше, чим більш
точно обчислено значення
.
Обчислення стандартного відхилення.
Одиничні
вимірювання в серії (вибірці)
характеризуються середньоквадратичною
помилкою або стандартним відхиленням
(![]() ):
):
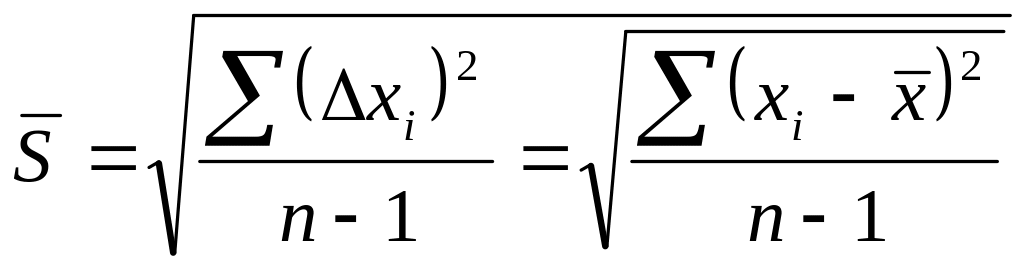 .
.
Обчислення середньоквадратичної помилки середнього.
Число
вибірок може бути скільки завгодно
великим. Кожна з них є випадковою зі
своєю середньою та середньоквадратичною
помилкою (![]() ).
).
 ;
;
Обчислення погрішності результатів вимірювань.
Обчислення погрішності результату вимірювання проводять при визначенні ступеня надійності α (частка випадків, при яких при даної кількості числа вимірювань середнє арифметичне лежить у певному інтервалі.
У
більшості випадків
![]() або
або
![]() .
Тобто 95% або 99% всіх вимірювань знаходяться
в указаних інтервалах.
.
Тобто 95% або 99% всіх вимірювань знаходяться
в указаних інтервалах.
![]()
Коефіцієнт
![]() називається коефіцієнтом нормованих
відхилень, або критерієм Стьюдента (К).
називається коефіцієнтом нормованих
відхилень, або критерієм Стьюдента (К).
Цю величину знаходять за спеціальними таблицями залежно від α та К = n – 1.
Величина К називається числом степеней вільності. Коефіцієнти нормальних відхилень для значень α, рівні 0,95 та 0,99, наведені в таблиці:
α |
К |
||||||||
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
|
0,95 |
4,30 |
3,18 |
2,78 |
2,57 |
2,45 |
2,37 |
2,31 |
2,26 |
2,23 |
0,99 |
9,93 |
5,84 |
4,60 |
4,03 |
3,71 |
3,50 |
3,36 |
3,25 |
3,17 |
Кінцевий результат вимірювань.
![]()
Обчислення відносної помилки.
![]()
Приклад №2
Для серії значень об’ємів титранту, що дорівнює 9,22; 9,26; 9,24; та 9,27мл, обчислити середнє значення та доверительний інтервал середнього при P=0,95.
Розв’язок:
Середнє значення дорівнює 9,248:
![]()
Границі відхилення:
![]()
![]()
![]()
![]()
Стандартне відхилення дорівнює 0,0222 мл:
![]()
![]()
![]()
Табличне значення коефіцієнта Стьюдента t(P=0,95; f = 3) = 3,18.
Доверительний інтервал дорівнює:
= 3,18 0,0222 = 0,035
![]() =
9,248 ±
0,035 = 9,25 ±
0,04мл
=
9,248 ±
0,035 = 9,25 ±
0,04мл
(отриманий результат заокруглюємо так, щоб напівширина доверительного інтервалу містила тільки одну значущу цифру).