

7. Теория переходного состояния.
Теория активных столкновений не в состоянии объяснить аномально медленное течение ряда реакций. Это объясняется тем, что она ограничивается чисто механическим рассмотрением столкновений молекул и не учитывает возможность последних участвовать во вращательном и колебательном движениях.
Кроме того, из термодинамических соображений следует, что стерический фактор должен быть связан с изменением энтропии в ходе химического превращения, так как меняется конфигурация размещения молекул в пространстве, что тоже не объясняет теория активных столкновений.
Развитию теории переходного состояния, называемой еще теорией активного комплекса, положили работы Эйринга и Поляни (1935 г.), в которых использованы основные представления теории активных столкновений и необходимость преодоления энергетического барьера в ходе химического превращения.
Основные положения теории: всякая химическая реакция протекает через образование некоторого активного комплекса. который затем распадается с образованием конечных продуктов химической реакции.
Так, например, реакцию А + ВС = АВ + С можно представить следующим образом:
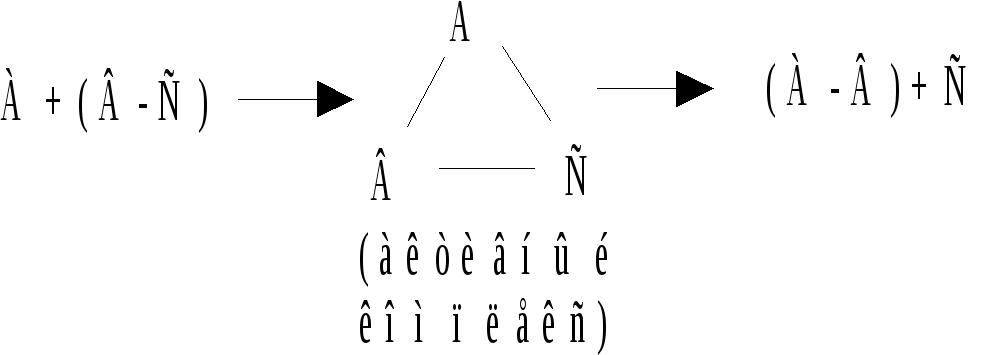
Следовательно, в ходе этой реакции реагирующие частицы образуют вначале некоторый малоустойчивый комплекс атомов А, В и С, который распадается на частицы конечных продуктов реакции.
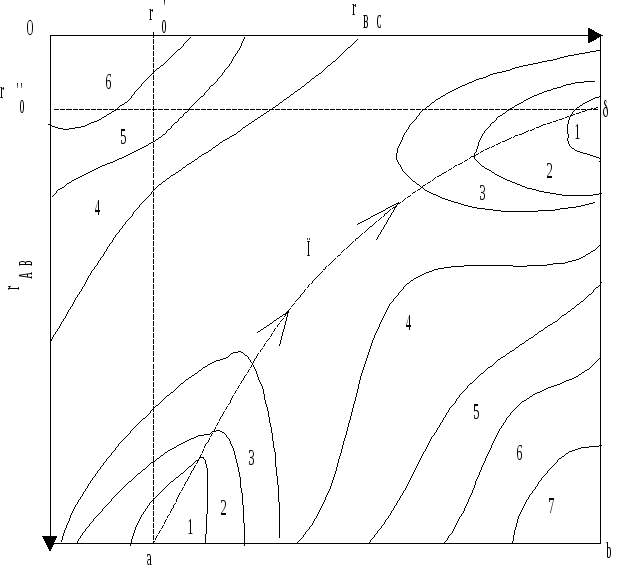
Современная физика позволяет оценить энергию реагирующей системы (W) как функцию расстояний между атомами (rAB и rBC). Так как энергия зависит от двух переменных, то ее изменение изображается в трехмерном пространстве W - rAB - rBС. Проекция этой диаграммы на плоскость с координатами rAB и rBC имеет вид, представленный рисунком 10.3.
Рис. 10.3. Энергетическая
диаграмма.
![]() ,
а rBС = r0’,
характеризующее расстояние между
центрами атомов В и С в молекуле ВС.
,
а rBС = r0’,
характеризующее расстояние между
центрами атомов В и С в молекуле ВС.
Конечное
состояние системы
(т.![]() ):
атом С и молекула АВ, то есть rBС
=
):
атом С и молекула АВ, то есть rBС
=
![]() ,
а rАВ
= r0”.
Величина r0”
- расстояние между центрами атомов А и
В в молекуле АВ.
,
а rАВ
= r0”.
Величина r0”
- расстояние между центрами атомов А и
В в молекуле АВ.
Точка b соответствует состоянию системы с разообщенными атомами А, В, С, а точка П состоянию, когда все три атома сближены и образуют как бы единую молекулу - активный комплекс.
Линии, нанесенные на диаграмму и обозначенные цифрами - изоэнергетические линии, поэтому исходное и конечное состояния находятся в энергетических “долинах”, а точка b - на энергетическом “плато”.
В процессе
химической реакции система из трех
атомов должна перейти из состояния т.
а в состояние т.![]() .
Наиболее выгодный путь с наименьшими
энергозатратами обозначен пунктирной
линией - координатой реакции.
.
Наиболее выгодный путь с наименьшими
энергозатратами обозначен пунктирной
линией - координатой реакции.
На этом пути имеется энергетический “перевал” - точка П, определяющая энергию образования активного комплекса или переходного состояния, для которого rAB = rBС. В переходном состоянии система обладает максимальной потенциальной энергией на наиболее выгодном пути реакции. Эта максимальная энергия и есть энергия активации химической реакции.
Таким образом, чтобы реакция произошла, энергия реагирующей системы должна позволить образоваться переходному состоянию. Вероятность осуществления химической реакции связывается с вероятностью образования переходного состояния, что открывает путь использования статистических методов для расчета скорости химической реакции.
В разработанной Эйрингом и Поляни теории переходного состояния принимается, что исходные продукты химической реакции находятся в равновесии с активными комплексами. Поэтому переходное состояние можно рассматривать как обыкновенную молекулу, имеющую кроме обычных трех степеней свободы поступательного движения четвертую степень свободы, связанную с движением вдоль пути (координаты) реакции.
Для рассмотренной выше реакции вида:
А + ВС = АВ + С
скорость реакции
прямо пропорциональна произведению
средней линейной концентрации
![]() активных компонентов на среднюю скорость
их перемещения
активных компонентов на среднюю скорость
их перемещения
![]() вдоль координаты реакции:
вдоль координаты реакции:
![]() ,
(10.32)
,
(10.32)
где
![]() - трансмиссионный коэффициент, учитывающий
вероятность распада комплекса на ко-
- трансмиссионный коэффициент, учитывающий
вероятность распада комплекса на ко-
нечные продукты.
Из (10.32) следует:
![]() ,
(10.33)
,
(10.33)
где
![]() - константа равновесия реакции образования
активного комплекса.
- константа равновесия реакции образования
активного комплекса.
Из молекулярно-кинетической теории следует:
![]() ,
(10.34)
,
(10.34)
где k - постоянная Больцмана;
h = 6,626
![]() 10-34
Дж
10-34
Дж![]() с,
постоянная Планка.
с,
постоянная Планка.
Поэтому:
![]() ,
(10.35)
,
(10.35)
то есть константа скорости реакции образования активного комплекса пропорциональна константе равновесия реакции его образования.
На основе термодинамического метода:
![]() ,
(10.36)
,
(10.36)
где
![]() - изменение энергии Гиббса при
образовании активного комплекса в
стандарт-
- изменение энергии Гиббса при
образовании активного комплекса в
стандарт-
ном состоянии.
Учитывая, что
![]() ,
окончательно:
,
окончательно:
![]() ,
(10.37)
,
(10.37)
где
![]() и
и
![]() - изменения энтропии и энтальпии при
образовании активного комплекса в
- изменения энтропии и энтальпии при
образовании активного комплекса в
стандартных условиях.
Уравнение (10.35) с учетом (10.37) примет вид:
![]() .
(10.38)
.
(10.38)
Из уравнения
(10.38) следует, что скорость химической
реакции определяется изменением энергии
Гиббса или Гельмгольца при переходе
молекул исходных продуктов реакции в
активный комплекс. Энергия активации
в теории переходного состояния заменяется
изменением энтальпии
![]() ,
а энтропийный сомножитель
,
а энтропийный сомножитель
![]() - энтропия активации,
тесно связана со строением исходных
молекул и активного комплекса. Например,
разрушение сложных молекул и образование
более простого активного комплекса
ведет к росту “беспорядка” в системе
и сопровождается повышением энтропии.
- энтропия активации,
тесно связана со строением исходных
молекул и активного комплекса. Например,
разрушение сложных молекул и образование
более простого активного комплекса
ведет к росту “беспорядка” в системе
и сопровождается повышением энтропии.
Зависимость скорости реакции не только от энергии активации, но и от энтропии активации, позволяет объяснить существование медленных реакций , имеющих малую энергию активации. быстрых реакций с большой энергией активации, различие скоростей реакции с одинаковыми энергиями активации.
Теория активного комплекса применима к реакциям, протекающим в растворах, тогда как теория столкновений хорошо описывает только реакции, протекающие в газовой фазе. Дело в том, что молекулы реагентов в жидкости находятся на более близком расстоянии, когда силы взаимодействия между ними нельзя считать малыми или даже отсутствующими, что часто допустимо в газах. В некоторых случаях растворитель не играет значительной роли, а в других, наоборот, сильно влияет на скорость реакции. Скорости реакций в растворах могут сильно отличаться от рассчитанных по теории активных столкновений как в ту, так и в другую сторону. Стерический фактор при этом может быть больше единицы как в реакциях между заряженными частицами, так и много меньше.