

8.3. Хромосомные болезни. Связь хромосомного дисбаланса с отклонениями в развитии.
Хромосомные болезни
К этой группе относят заболевания, вызванные аномалиями числа или структуры хромосом. Около 1 % новорожденных имеют аномальный кариотип, а среди мертворожденных встречаемость аберраций числа или структуры хромосом – 20 %. Общими характерными чертами хромосомных болезней являются: низкий вес при рождении, задержка развития, низкий рост, микроцефалия, микрогнатия, нарушения остеогенеза, аномальное расположение глаз.
Синдром «кошачьего крика» был описан в 1963 г. Дж.Леженом. Частота его среди новорожденных составляет 1:45 000. Причиной данного заболевания является делеция части короткого плеча 5-й хромосомы. Показано, что лишь небольшой участок короткого плеча хромосомы-5 ответственен за развитие полного клинического синдрома..
|
Наиболее характерным симптомом этого заболевания является специфический плач новорожденных, похожий на кошачий крик. Возникновение специфического крика связано с изменениями гортани — сужением, мягкостью хрящей, отечностью или необычной складчатостью слизистой, уменьшением надгортанника. У этих детей часто выявляются микроцефалия, низко расположенные и деформированные ушные раковины, микрогения, лунообразное лицо, гипертелоризм, эпикант, монголоидный разрез глаз и мышечная гипотония. Дети резко отстают в физическом и умственном развитии Такие диагностические признаки, как «кошачий крик», лунообразное лицо и гипотония мышц, с возрастом исчезают полностью, а микроцефалия, напротив, становится более очевидной, прогрессирует и умственная отсталость (рис.13). |
|
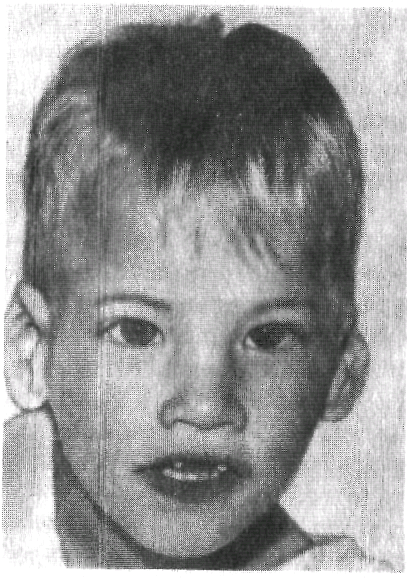
Рис. 13 Синдром кошачьего крика
8.4. Геномные болезни
Наиболее распространенный случай анеуплоидии – трисомия (наличие одной добавочной хромосомы) по хромосоме 21 или синдром Дауна. Эта аномалия встречается с частотой 1 на 700 новорожденных. Риск рождения ребенка с синдромом Дауна повышается с увеличением возраста матери. Для больных характерны признаки: брахицефалия, уплощенный затылок, скошенный и узкий лоб, плоское лицо, эпикант, монголоидный разрез глаз, короткий нос, постоянно открытый рот, толстые губы, большой складчатый язык (рис.14). Ушные раковины уменьшены и деформированы. Со стороны костно-мышечной системы характерны низкий рост, короткая шея, килевидная или воронкообразная деформация грудины, мышечная гипотония (особенно выраженная у маленьких детей), широкие киста и стопы с короткими пальцами, поперечная ладонная складка, двухфаланговый мизинец. Примерно в 50 % случаев у детей отмечаются различные пороки сердца — дефекты межжелудочковой и межпредсердной перегородок, аномалии крупных сосудов. Наиболее характерны атрезия или стеноз двенадцатиперстной кишки, несколько реже встречаются атрезии прямой кишки и ануса, а также пищевода. У больных отмечается задержка роста и развития, умственная отсталость, врожденный порок сердца, снижение иммунитета, снижение мышечного тонуса. Средняя продолжительность жизни – 49 лет. Больные мужчины бесплодны, у некоторых женщин могут быть дети как с синдромом Дауна, так и нормальные. Запись кариотипа с трисомией по хромосоме 21 выглядит так: 47, ХХ, +21 или 47, ХY, +21. Кроме дополнительной хромосомы 21 причиной этого синдрома могут быть внутрихромосомные перестройки с ее участием.

Рис. 14 Ребенок с синдромом Дауна
Генетическая природа синдрома Патау, трисомия 13, была расшифрована в 1960 г. американским генетиком К. Патау, чьим именем в дальнейшем он и был назван. Частота данного заболевания составляет 1 на 6000 рождений, занимая второе место по частоте встречаемости (после синдрома Дауна) среди полных аутосомных трисомий. Простая полная трисомия 13 (47, XX (XY) + 13) как следствие нерасхождения этой пары хромосом в мейозе у одного из родителей (главным образом у матери) встречается в 80-85% случаев заболевания (рис.15). Остальные случаи обусловлены транслокациями. Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией. Особенно обращают на себя внимание аномалии черепа и лица - микроцефалия, в ряде случаев отмечается выраженная тригоноцефалия, скошенный лоб, узкие глазные щели, гипотелоризм, запавшее переносье, низкорасположенные и деформированные ушные раковины. Наиболее характерными внешними пороками развития являются расщелина губы и нёба и полидактилия (рис.). Врожденные пороки сердца отмечаются у 80% детей. Пороки пищеварительного тракта отмечаются у половины больных. Пороки развития почек наблюдаются в 60% случаев, наиболее характерным является поликистоз. Пороки развития органов зрения — анофтальмия, микрофтальмия, дисплазии сетчатки, колобома радужки, помутнение хрусталика — встречаются более чем у 70% больных. Центральная нервная система поражается в 100% случаев.

Рис. 15 Синдром Патау
Продолжительность жизни у детей с синдромом Патау резко снижена. На первом году жизни умирают 95% больных, причем 60-65% в перинатальном периоде. В возрасте старше 3 лет остаются в живых единицы. Все дети с синдромом Патау имеют тяжелую умственную отсталость.
Для синдрома Эдвардса характерно сочетание специфических клинических проявлений: долихоцефалия, гипоплазия нижней челюсти и микростомия, узкие и короткие глазные щели, маленькие низко расположенные ушные раковины, характерное сгибательное положение пальцев кисти, выступающий затылок и другие микроаномалии (рис.16). При синдроме практически постоянны пороки сердца и крупных сосудов, часты пороки желудочно-кишечного тракта, пороки почек и половых органов. Продолжительность жизни больных с синдромом Эдвардса резко снижена. На первом году жизни погибают 90% больных, к 3-летнему возрасту — более 95%. Причиной смерти являются пороки сердечно-сосудистой системы, кишечника или почек. Все выжившие больные имеют глубокую степень олигофрении (идиотию).

Рис. 16 Синдром Эдвардса
Синдром Шерешевского-Тернера
Анеуплоидии по половым хромосомам также достаточно часто встречаются в популяциях человека. Моносомия (отсутствие одной из гомологичных хромосом) по Х-хромосоме или синдром Шерешевского-Тёрнера встречается с частотой 1 на 1500.
Кариотип – 45, Х. Пол ребенка с моносомией по Х-хромосоме женский, так как у всех млекопитающих мужской пол определяется наличием Y-хромосомы, а не количеством Х-хромосом. Вынашивание девочек с этим синдромом часто проходит с токсикозом и угрозой выкидыша, а роды бывают преждевременными и патологическими. Во второй половине беременности происходит инволюция (обратное развитие) половых клеток, и к моменту рождения у ребенка резко уменьшено количество фолликулов или они вовсе отсутствуют.
Следствиями недоразвития гонад являются недостаточность женских гормонов, аменорея (отсутствие менструаций) и бесплодие. Для больных девочек характерен низкий рост, отставание в развитии, широкая бочкообразная грудная клетка, короткая шея, крыловидные складки на боковых поверхностях шеи, деформация локтевых суставов (рис.17). Часто встречаются пороки сердца и крупных сосудов, аплазия фаланг пальцев, склонность к ожирению и гипертензия.
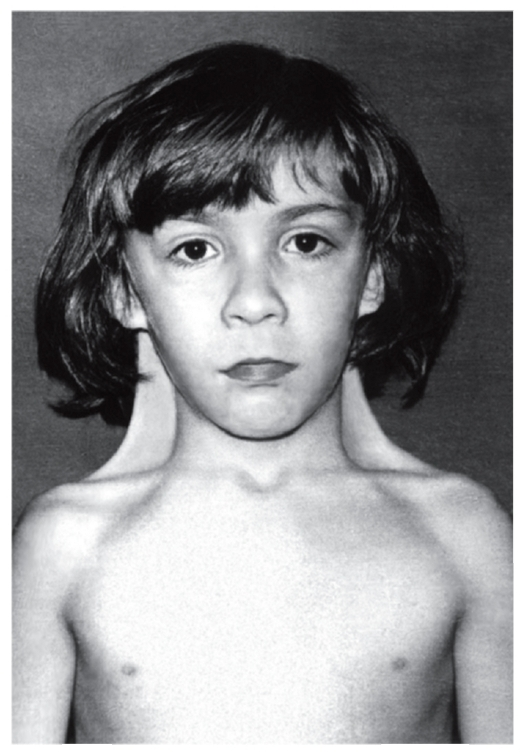
Рис. 17 Синдром Шерешевского-Тёрнера
Синдром тройной Х-хромосомы встречается с частотой 1 на 700. Все больные – внешне нормальные женщины с кариотипом 47, ХХХ. Иногда встречаются две или более дополнительные Х-хромосомы. Умственная отсталость и алалия отмечаются у 75 % больных, часто наблюдается недоразвитие фолликулов, ранний климакс и бесплодие.
Синдром Клайнфельтера и органический инфантилизм по типу психической неустойчивости.
Одна или несколько дополнительных Х-хромосом у представителей мужского пола – синдром Клайнфельтера. Возможны кариотипы: 47, XXY; 48, XXYY; 48, XXXY; 49, XXXXY; 49, XXXYY). Частота встречаемости 1 на 500–700 новорожденных мальчиков. Для больных характерны высокий рост, длинные конечности при сравнительно коротком туловище, гинекомастия, евнухоидизм, бесплодие, повышенное содержание женских гормонов, ожирение, психические нарушения, склонность к асоциальному поведению (рис.). Отклонения в физическом развитии проявляются в препубертате и пубертате. У больных отмечается евнухоидное телосложение, в 25 % случаев — гинекомастия (увеличение грудных желез) и высокий рост (рис.18).
Наружные половые органы сформированы по мужскому типу, размеры полового члена нормальные, яички резко уменьшены в размерах. Именно микроорхидизм является главным клиническим признаком этого синдрома. Объем эякулята снижен, выявляется азооспермия. Мужчины с этим синдромом бесплодны.

Рис. 18 Синдром Кляйнфельтера
Синдром дополнительной Y-хромосомы (кариотип 47, XYY) встречается с частотой 1 на 500. Клинические проявления практически не выражены. Часто встречаются высокий рост, атлетическое телосложение, несколько повышенный уровень агрессивности, неадекватная реакция на критику, склонность к импульсивным поступкам, любовь к риску и приключениям. Синдром с большей, чем в среднем по популяции, частотой встречается среди осужденных за насильственные преступления, представителей опасных профессий и любителей экстремальных видов спорта.