

8.2.2. Нарушения липидного обмена
Болезнь Ниманна-Пика типов А и Б – снижение активности фермента кислой лизосомальной сфингомиелиназы, который кодируется геном SMPD1. Тип наследования – аутосомно-рецессивный. Нарушение липидного метаболизма приводит к накоплению липидов в печени, легких, селезенке, нервных тканях. Характерна дегенерация нервных клеток, нарушение деятельности нервной системы, повышенный уровень холестерина и липидов в крови. Тип А летален в раннем детском возрасте. Тип Б протекает более мягко, больные, как правило, доживают до взрослого состояния. (рис. 7).

Рис. 7 Болезнь Ниманна-Пика
Болезнь Гоше (гликозилцерамидный липидоз) – накопление глюкоцереброзидов в клетках нервной и ретикулоэндотелиальной системы, обусловленное дефицитом фермента глюкоцереброзидазы, которая кодируется геном GBA. Относится к группе лизосомных болезней накопления. Некоторые формы заболевания проявляются в тяжелых поражениях печени, селезенки, нервной и костной тканей (рис. 8).

Рис. 8 Болезнь Гоше
Наследственные болезни пуринового и пиримидинового обмена
Синдром Леша-Нихена – сцепленное с полом рецессивное заболевание, при котором резко возрастает содержание мочевой кислоты во всех жидкостях тела. Последствием этого является задержка развития, умеренная умственная отсталость, приступы агрессивного поведения с самоповреждением. Недостаточность ферментативной активности гипоксантин гуанинфосфорибозилтрансферазы по причине мутаций в гене HPRT1 лежит в основе этого заболевания. Описаны несколько мутаций в том же гене, следствием которых является подагра (нарушение пуринового обмена и отложение мочекислых соединений в тканях) (рис.9).
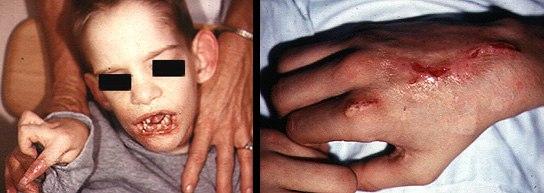
Рис. 9 Синдром Леша-Нихена
8.2.3. Нарушения обмена соединительной ткани
Синдром Марфана («паучьи пальцы», арахнодактилия) – поражение соединительной ткани вследствие мутации в гене FBN1, ответственном за синтез фибриллина. Наследуется по аутосомно-доминантному типу. Клиническая полиморфность заболевания объясняется большим числом мутантных аллелей, каждый из которых может проявляться в гетерозиготном состоянии. Для больных характерен высокий рост, астеническое телосложение (непропорционально длинные конечности), арахнодактилия (длинные тонкие пальцы), слабость связочного аппарата, отслойка сетчатки глаза, подвывих хрусталика, пролапс митрального клапана (рис.10).

Рис. 10 Синдром Марфана
Мукополисахаридозы – группа заболеваний соединительной ткани, связанных с нарушеним обмена кислых гликозаминогликанов (мукополисахаридов), вызванных недостаточностью некоторых лизосомных ферментов. Эти заболевания относят к лизосомным болезням накопления. Они проявляются в различных дефектах костной и соединительной тканей. Мукополисазаридоз типа I (синдром Хурлера) – аутосомно-рецессивное заболевание, возникающее в результате дефицита фермента альфа-L-идуронидазы из-за мутаций в гене IDUA. Это приводит к накоплению белково-углеводных комплексов и жиров в клетках организма. В результате у больных наблюдается малый рост, существенная задержка умственного развития, увеличение печени и селезенки, пороки сердца, помутнение роговицы, деформация костей и огрубение черт лица.
Мукополисахаридоз типа II (синдром Хантера) – сцепленное с полом рецессивное заболевание, которое обусловлено дефектом фермента идуронатсульфотазы из-за мутации в гене IDS. Веществами накопления являются дерматан- и гепарансульфаты. Характерны грубые черты лица, скафоцефалия, шумное дыхание, низкий грубый голос, частые острые респираторные вирусные инфекции (рис.11). Наблюдаются прогрессирующая тугоухость, узелковые поражения кожи спины, остеоартриты, поражения роговицы.
\

Рис. 11 Синдром Хантера
Мукополисахаридоз типа III (синдром Санфилиппо, болезнь Санфилиппо) – заболевание, вызванное накоплением гепарансульфата. Для него характерна генетическая гетерогенность – существуют четыре типа этой болезни, вызванные мутациями в четырех разных генах, кодирующих ферменты, участвующие в метаболизме накапливаемого вещества.. Для больных арактерны задержка роста, контрактуры суставов, гипертрихоз, умеренная гепатоспленомегалия. В отличие от синдромов Хурлера и Хантера при болезни Санфилиппо преобладает умственная отсталость (рис.12).

Рис. 12 Мукополисахаридоз
Особенности патогенеза моногенных болезней
Мутационное изменение нуклеотидной последовательности структуры ДНК является причиной моногенных наследственных болезней. Специфика патогенеза моногенных заболеваний определяется особенностями химической природы первичного продукта, обусловленными конкретной мутацией, и той ролью, которую этот продукт играет в жизнедеятельности организма. При одних мутациях, обусловливающих полное отсутствие необходимого организму вещества (например, соматотропного гормона или цитохрома-450), нормальное развитие организма либо затруднено, либо невозможно; при других мутациях, приводящих к дефициту биологически активного вещества или структурного белка, возникают заболевания, характеризующиеся расстройством структуры и функции отдельных тканей, органов или физиологических систем.
К настоящему времени открыты и подробно описаны сотни видов наследственных аномалий метаболизма, причина которых единичный мутантный ген. Обусловленная мутацией гена аномалия аминокислотной последовательности полипептидной цепи существенно нарушает активность ферментов за счет изменений количества белка, его информационных характеристик, термостабильности, устойчивости к различным воздействиям и других свойств.