

РАЗДЕЛ ГЕНЕТИКА И МЕДИЦИНА
Тема 8 Наследственные патологии
8.1. Понятие, классификация и особенности наследственной патологии
Патология – это любое отклонение от нормального течения биологических процессов – обмена веществ, роста, развития, размножения. Наследственная патология – отклонение от нормы с установленным фактом наследования, т. е. передачи от поколения к поколению. Следует различать врожденную патологию – присутствующую от рождения индивидуума – от наследственной патологии. Врожденная патология может быть обусловлена действием факторов внешней среды – недостатком питательных веществ и кислорода во время внутриутробного развития, родовыми травмами, инфекциями и т. д. Установление в соответствии с требованиями генетического анализа факта наследования аномального признака является единственным основанием признания наследственного характера патологии.
8.2. Моногенные болезни. Характеристика отдельных форм.
Генными болезнями называют патологические состояния, причиной которых являются генные мутации. Для этой группы характерна гетерогенность – одинаковые заболевания могут быть вызваны мутациями в разных генах.
Общими принципами развития патологии на уровне генов могут быть следующие:
- выработка аномального белкового продукта;
- отсутствие нормального белка;
- недостаточное количество нормального белка;
- избыток нормального белкового продукта.
8.2.1. Болезни аминокислотного обмена
Самая многочисленная группа наследственных болезней обмена веществ. Почти все они наследуются по аутосомно-рецессивному типу. Причина заболеваний – недостаточность того или иного фермента, ответственного за синтез аминокислот.
Фенилкетонурия – нарушение превращения фенилаланина в тирозин из-за резкого снижения активности фенилаланингидроксилазы – аутосомно-рецессивное заболевание. Проявляется в возрасте 2–4 месяцев, первые симптомы – вялость, судороги, экзема, «мышиный» запах (запах кетонов). Постепенно развиваются тяжелые поражения головного мозга, приводящие к резкому снижению интеллекта вплоть до идиотии. Если с первых дней жизни полностью исключить (или существенно ограничить количество) фениаланин из рациона больного ребенка до полового созревания, симптомы не развиваются. Болезнь обусловлена мутациями в гене PAH, который кодирует фенилаланин-4-гидроксилазу.

Рис. 1. Фенилкетонурия
Алкаптонурия – аутосомно-рецессивное нарушение обмена тирозина и накопления в тканях организма (суставные хрящи, сухожилия) гомогентизиновой кислоты. Манифестация происходит в детском возрасте. Первый симптом – потемнение мочи. Часто развивается мочекаменная болезнь и пиелонефрит. Накопление продуктов распада гомогентизиновой кислоты приводит к поражению суставов (в первую очередь коленных и тазобедренных). Отмечается потемнение и повышенная хрупкость соединительной ткани. Характерно потемнение склер и ушных раковин.

Рис. 2 Алкаптонурия
Глазо-кожный альбинизм первого типа – отсутствие или существенный недостаток пигмента кожи, волос, радужной и пигментной оболочек глаза (рис.3). Заболевание с аутосомно-рецессивным типом наследования. Проявляется в различной степени депигментации кожи, волос, радужной и пигментной оболочек глаза, снижением остроты зрения, светобоязни, нистагме, частых солнечных ожогах. Различные миссенс-мутации, мутации типа сдвига рамки считывания и нонсенс мутации в гене тирозиназы.
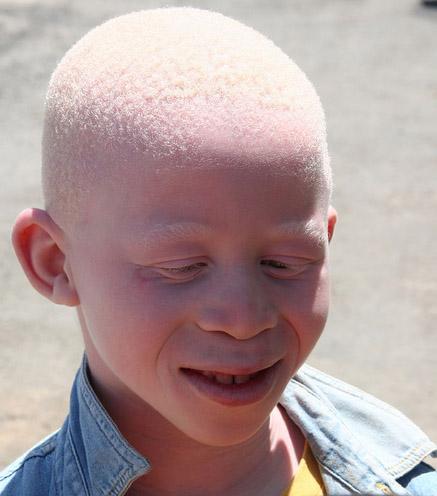
Рис. 3 Альбинизм
Гомоцистинурия
Минимальные диагностические признаки: марфаноидный фенотип, повышение концентрации метионина и гомоцистина в плазме крови в сочетании со снижением того же показателя для цистина, а также повышением экскреции гомоцистина с мочой (гомоцистинурией) (рис. 4).
В основе патогенеза гомоцистинурии лежит нарушение обмена серосодержащих аминокислот. В норме одна из таких аминокислот — метионин — через ряд промежуточных стадий (в том числе гомо-цистеин и цистатионин) переходит в цистеин.
Повышенная концентрация этих естественных для организма веществ вызывает очаговые некрозы в почках, селезенке, слизистой оболочке желудка, сосудах. Повреждения стенок сосудов, а также активизация свертывающей системы крови усиливают тромбообразование, что проявляется тромбозами коронарных, сонных, почечных артерий, генерализованным венозным тромбозом. Это может приводить к артериальной гипертензии, различным неврологическим нарушениям, ранней смерти.

Рис. 4 Гомоцистинурия
Галактоземия – отсутствие или существенное снижение активности фермента галактозо-1-фосфат-уридилтрансферазы и накопление в крови галактозы и ее производных, которые оказывают токсическое действие на центральную нервную систему, печень и хрусталик глаза. В первые дни и недели жизни наблюдаются желтуха, увеличение печени, нистагм, гипотония мышц, рвота. Со временем развивается катаракта, отставание в физическом и умственном развитии. Характерна непереносимость молока. Болезнь имеет аутосомно-рецессивный тип наследования. Несколько форм этого заболевания обусловлены различными мутантными аллелями гена галактозо-1- фосфатуридилтрансферазы (рис. 5).

Рис. 5 Галактоземия
Болезнь Гирке (гликогеноз I типа, гликогеновая болезнь I типа) – неспособность превращения глюкозо-6-фосфата в глюкозу, которая приводит к нарушению как синтеза, так и разложения гликогена. Депонирование гликогена происходит, обратный процесс – нет. Развивается гипогликемия. Накопление избыточного количества гликогена в печени и почках приводит к печеночной и почечной недостаточности. Тип наследования – аутосомно-рецессивный. Причина заболевания – мутация в гене, который кодирует фермент глюкозо-6-фосфатазу. Описано 14 мутантных аллелей этого гена (рис. 6).
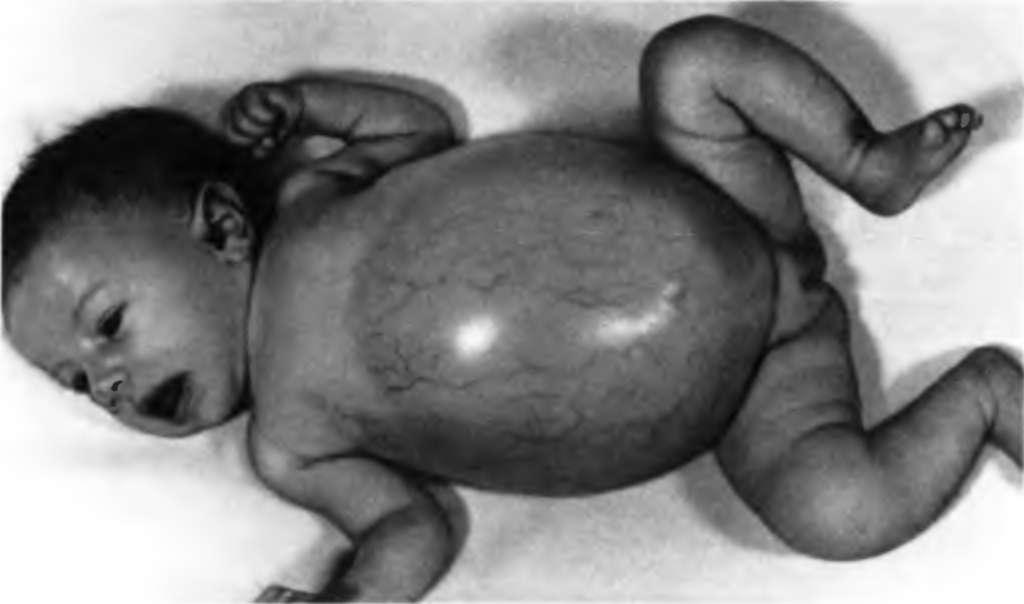
Рис. 6 Болезнь Гирке