

Reactive Intermediate Chemistry
.pdf132 RADICALS
ENDOR spectroscopy has two advantages over ESR spectroscopy. The simplicity of the ENDOR spectrum is obvious. For the triarylmethyl radical in Figure 4.4, the ENDOR spectrum25 consists of six lines (the central line in the figure from coupling to the tert-butyl protons is actually two closely spaced lines), whereas the ESR spectrum has 28 well-resolved lines. A second advantage of ENDOR spectroscopy is that identification of the nuclei coupled to the unpaired electron is simple. For example, the a values for a 13C atom at a carbon radical center and an a proton are similar, but the radio wave frequencies at which 13C and 1H absorb in a given magnetic field are tremendously different (e.g., 75 vs. 300 MHz). This feature makes ENDOR spectroscopy especially attractive for identifying the types of the atoms coupled to the unpaired electron, which, for example, is useful in characterizing large, complex biological radicals.
2.5. Chemically Induced Dynamic Nuclear Polarization Effects
Chemically induced dynamic nuclear polarization (CIDNP; pronounced sid-nip or kid-nip) is a phenomenon first reported in the 1960s, which involves the observation of strongly enhanced absorbances and strong emissions in NMR spectra obtained from solutions in which a radical reaction is occurring. The CIDNP is a misnomer based on the apparent similarity of the effect to those seen in dynamic nuclear polarizations, where one pumps electron spins and observes NMR signals. An
operational theory explaining CIDNP was expounded soon after early reports of the effect,28,29 and it is known as the radical pair theory. The effect involves the
phenomena described below in the context of a reaction that initially gives a singlet radical pair, but triplet radical pairs formed in photolysis reactions also can be explained.
Assume that a homolysis reaction is being conducted in a tube in an NMR spectrometer. A singlet radical pair is formed by homolysis, and this pair can recombine, diffuse apart, or undergo intersystem crossing (ISC, an electron spin flip) to give a triplet pair. The triplet pair thus formed can diffuse apart or return to the singlet pair by ISC, but it cannot recombine because that would produce a highenergy triplet state of a closed-shell molecule. Because diffusional processes have the same rates for both types of pairs, the relative amount of escape from the triplet pair is greater than that from the singlet pair.
The radicals formed in the homolysis maintain the nuclear spin character of their protons during the lifetime of the radical pair. Differences in proton nuclear spin give slightly different energy levels for the radicals in a magnetic field. This phenomenon is the same effect as seen in hyperfine coupling that gives multiplets in the ESR spectrum, but the magnetic field in this experiment is that of the NMR spectrometer. The rates of ISC differ slightly for different energy sublevels of the radical pairs and are thus a function of the nuclear spins of the protons. Therefore, the singlet and triplet radical pairs develop non-Boltzmann proton nuclear spin distributions. Further, diffusive free radicals that escape from the radical pairs also will have non-Boltzmann proton nuclear spin distributions.
A radical that escapes from a radical pair can react with a molecule to give a closed-shell product and a new radical, or it can react with another radical to
IDENTIFICATION AND CHARACTERIZATION OF RADICALS |
133 |
give a new radical pair. A closed-shell product also can be formed by recombination of radicals in the original or new singlet radical pair. In both types of reactions, the closed-shell products will have non-Boltzmann proton nuclear spin populations, and it is these products that produce the CIDNP effects in NMR spectra. The nuclear spins will relax thermally, but, if the NMR spectrum is obtained before thermal relaxation is complete, one will observe large enhanced absorption or emission from the product molecules.
CIDNP effects can be seen when a reaction that gives a radical pair is reasonably fast, with a lifetime on the order of minutes. The small differences in highand lowenergy nuclear spin states that exist in the magnetic field of the NMR spectrometer, giving nearly equal populations of the spin states when thermally equilibrated, and the extremely high sensitivity of an NMR instrument are the features that combine to make CIDNP effects observable. The NMR spectrometer reports on differences in nuclear spin state populations of parts per million, and, for example, only a small fraction of nuclear-spin excited molecules are needed to obtain an emission signal. The high sensitivity of the method can be a problem, however, because it is possible to observe CIDNP signals originating from minor byproducts of the reaction. This is seen in the example in Figure 4.4 where CIDNP signals from 1-butene (at d 5.6 and 6.1) are apparent, but those NMR signals are not observed in the spectrum of the product.
The spectra shown in Figure 4.4 were obtained from a reaction of tert-butyl- lithium with 1-bromobutane.30 One deduces that both the tert-butyl and the butyl radical were produced and that they reacted in disproportionation reactions to give in part isobutylene and 1-butene, respectively. In the spectrum recorded at 30 s, vinyl proton signals from 1-butene are in emission (d 5.0 and d 6.1) and enhanced absorption (d 5.6). The isobutylene vinyl proton signal at d 4.6 is in emission on the left side and enhanced absorption is on the right side. This phenomenon is known as a multiplet effect, and it is due to differences in ISC rates for radical pairs containing tert-butyl radicals with different proton nuclear spins. Note that the tert-butyl- lithium sample contained an impurity of isobutylene before the reaction, and the amount of isobutylene was increased after the reaction.
2.6. Absorption Spectroscopy
In principle, absorption spectroscopy techniques can be used to characterize radicals. The key issues are the sensitivity of the method, the concentrations of radicals that are produced, and the molar absorptivities of the radicals. High-energy electron beams in pulse radiolysis and ultraviolet–visible (UV–vis) light from lasers can produce relatively high radical concentrations in the 1–10 10 5 M range, and UV–vis spectroscopy is possible with sensitive photomultipliers. A compilation of absorption spectra for radicals contains many examples.31 Infrared (IR) spectroscopy can be used for select cases, such as carbonyl-containing radicals, but it is less useful than UV–vis spectroscopy. Time-resolved absorption spectroscopy is used for direct kinetic studies. Dynamic ESR spectroscopy also can be employed for kinetic studies,32,33 and this was the most important kinetic method available for reactions
134 RADICALS
in organic solvents before the introduction of lasers. Chapter 18 in this volume21b on kinetics in the nanosecond realm discusses the methods.
3. MULTISTEP RADICAL REACTIONS
Most radicals are highly reactive, and there are few examples where one would produce a stable radical product in a reaction. Reference to a ‘‘radical reaction’’ in synthesis or in Nature, almost always concerns a sequence of elementary reactions that give a composite reaction. Multistep radical sequences are discussed in general terms in this section so that the elementary radical reactions presented later can be viewed in the context of real conversions. The sequences can be either radical chain reactions or radical nonchain reactions. Most synthetic applications involve radical chain reactions, and these comprise the bulk of organic synthetic sequences and commercial applications. Nonchain reaction sequences are largely involved in radical reactions in biology. Some synthetic radical conversions are nonchain processes, and some recent advances in commercial polymerization reactions involve nonchain sequences.
3.1. Radical Chain Reactions
Radical chain reactions are comprised of three distinct parts: initiation, propagation steps, and termination. The initiation portion involves one or more elementary reactions that produce a radical that can participate in one of the propagation steps. The propagation sequence is where the desired products are formed; it consists of two or more reactions in which one product of each elementary reaction is a radical that serves as a reactant in another step of the sequence. Radicals are destroyed in termination steps that give nonradical products by radical–radical coupling and disproportionation reactions.
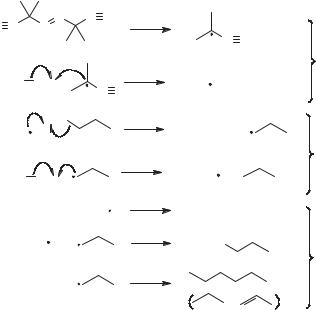
A simple radical chain reaction sequence is shown in Figure 4.6. This reaction sequence is an example of a ‘‘tin hydride reaction,’’ where an alkyl halide is reduced to an alkane. The initiation steps involve thermolysis of azobisisobutyrylnitrile (AIBN) to give the 1-cyano-1-methylethyl radical that reacts with tributyltin hydride (the most commonly used tin hydride) to give the tributylstannyl radical. In one propagation step, the tin radical reacts with the alkyl halide to give an alkyl radical and tributyltin bromide. In the second propagation step, the alkyl radical reacts with the tin hydride to give an alkane and another tributylstannyl radical. In this example, three types of radical–radical reactions can serve as termination steps in principle, but the most important termination reaction will involve selfreactions of the radical with the largest concentration as discussed in Section 3.2. The key to a successful chain reaction sequence is that the velocities of the propagation reactions are much greater than those of the termination reactions such that many cycles of the propagation sequence are completed for each radical initiation and termination reaction. To obtain the high velocities necessary in the propagation steps, the rate constants for these reactions must be reasonably large. In practice, it
|
|
|
|
MULTISTEP RADICAL REACTIONS 135 |
|
N C |
N N |
C N |
∆ |
|
|
|
|
|
|||
|
|
|
2 |
C N + N2 |
|
|
|
|
|
||
|
|
|
|
|
initiation |
Bu3Sn |
H |
C N |
Bu3Sn + |
(CH3)2CHCN |
|
|
|
||||
Bu Sn |
Br |
|
Bu3SnBr |
+ |
|
|
|
||||
3 |
|
|
|
|
|
|
|
|
|
|
propagation |
Bu Sn |
H |
|
Bu3Sn |
+ |
|
3 |
|
|
|
|
|
|
2 Bu3Sn |
|
Bu SnSnBu |
||
|
|
|
|
3 |
3 |
Bu3Sn + |
|
Bu3Sn |
termination |
||
|
|
|
|
|
|
2
+
Figure 4.6. Elementary processes in the radical chain reduction of an alkyl bromide.
is difficult to use a propagation reaction that has a pseudo-first-order rate constant <1 104 s 1 at ambient temperature.
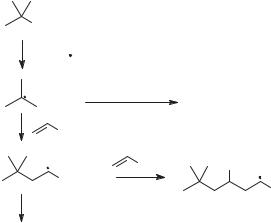
Additional elementary reactions can be incorporated into the propagation sequence to give more sophisticated synthetic conversions, but additional propagation steps inevitably produce increasing numbers of undesired reactions. For example, assume one wanted to prepare the addition product from the tert-butyl radical and methyl acrylate (Fig. 4.7). The desired propagation sequence now consists of three elementary reactions: bromine abstraction by the stannyl radical, addition of tert-butyl radical to the acrylate ester, and tin hydride trapping of the adduct radical. Two serious side reactions are now possible, reduction of the tert-butyl radical in competition with the addition to methyl acrylate, and addition of the adduct radical to a second molecule of acrylate in competition with the desired tin hydride reaction. Altering the concentration of one reagent to maximize a reaction in the desired sequence, such as decreasing the tin hydride concentration to avoid reduction of the alkyl radical, will reduce the efficiency of the other desired reaction. In practice, the conversion shown in Figure 4.7 cannot be made highly efficient because the rate constants for reaction of tin hydride with both types of radicals are essentially the same as are the rate constants for reaction of methyl acrylate with both types of radicals. If the newly added propagation reaction was a unimolecular reaction, such as a 5-exo radical cyclization, however, high yields of product could be obtained from the sequence halide reaction with stannyl radical, radical cyclization, and reaction of the cyclic radical with tin hydride.
136 RADICALS
Br |
|
Bu3Sn |
|
Bu3SnH |
Me2CHMe |
|
|
CO2Me |
|
CO Me |
CO2Me |
2 |
|
CO2Me |
CO2Me |
|
|
Bu3SnH |
|
 CO2Me desired product
CO2Me desired product
Figure 4.7. A three-step radical propagation sequence (top to bottom) that has two serious possible side reactions.
The number of undesired reactions increases rapidly as more steps are added to the propagation sequence with the result that achieving high-yield multistep conversions might seem unlikely. Nonetheless, many complex sequences can be achieved by controlling concentrations of reagents, incorporating unimolecular processes, and matching the polarities of radicals and radicophiles in the desired reaction sequence. For planning complex sequences, one needs absolute or relative kinetic information.
In radical chain polymerization reactions, the propagation sequence involves additions of a growing polymer chain to monomer molecules. In principle, each step is distinct, but, after the first addition step of the polymerization, subsequently formed polymeric radicals are similar in reactivity. To a good approximation, the polymerization reaction will behave as if there is a single propagation step. However, when the polymeric radicals grow large enough such that the reaction solution becomes viscous, termination reactions slow dramatically, and the concentration of radicals increases resulting in accelerated polymerization in the gel solution.34
3.2. Velocities of Radical Chain Reactions
Radical chain reactions are complicated because multiple reactions occur, but the overall velocity of the sequence can be given in simplified form by applying steadystate approximations. An important feature of any chain reaction is that the velocities of all propagation steps must be identical because the radicals formed as products in each elementary reaction are the reactants in another elementary
MULTISTEP RADICAL REACTIONS |
137 |
reaction. In addition, under steady-state conditions, the total velocity for radical termination reactions is equal to the velocity of radical initiation reactions.
The equality in the velocities of the chain propagation steps results in a relatively high concentration of the radical that reacts in the slowest chain propagation step. For example, for the tin hydride reduction of an alkyl bromide shown in Figure 4.6, the reaction of Bu3SnH with an alkyl radical has a rate constant of 2 106 M 1 s 1 at ambient temperature, and the rate constant for reaction of the tributylstannyl radical with 1-bromopropane is 3 107 M 1 s 1. If the initial concentrations of Bu3SnH and 1-bromopropane were equal, then the concentration of the propyl radical must be 15 times as great as the concentration of the stannyl radical for the velocities of the propagation steps to be equal. Because the termination steps in this radical sequence occur with similar (diffusion-controlled) rate constants, the most significant termination step will be the self-reaction of propyl radicals. Accordingly, the square of the propyl radical concentration is the concentration term that appears in the rate law for the termination reaction. The rate expression for the chain reaction is derived by setting the equality for velocities of initiation and termination and using the required equalities in the velocities of the propagation reactions. The derived rate law contains only one propagation rate constant, that for the ‘‘slow’’ propagation step. Specifically, for the tin hydride reduction in Figure 4.6, the rate law is
d½RH&=dt ¼ kSnH½Bu3SnH&ðFkln½AIBN&=2kTÞ1=2
where kSnH is the rate constant for the tin hydride reaction with the alkyl radical, F is the fraction of free radicals that escape from the solvent cage (between 0 and 2) when AIBN is thermolized, kIn is the rate constant for AIBN thermolysis, and kT is the diffusional rate constant for radical–radical reactions (kT is discussed in Section 4.3).
The slow radical propagation step in the chain sequence is termed the ratecontrolling step because its rate constant appears in the rate law for the chain reaction, and its importance is apparent. If one substituted 1-iodopropane for 1-bromo- propane in the reaction in Figure 4.6 and the concentrations and reaction conditions were otherwise unchanged, then the velocity of the chain reaction would not be altered. The steady-state concentration of the stannyl radical would be considerably reduced, however, from 7% of the concentration of the propyl radical in the bromopropane reduction to 0.1% of the concentration of the propyl radical in the iodopropane reduction. Alternatively, if the metal hydride reducing agent were changed to tris-(trimethylsilyl)silane, (Me3Si)3SiH, which reacts with the propyl radical with a rate constant 0.2 times that of tin hydride, then the velocity of the chain reaction would be reduced by a factor of 5.
Rate laws for radical chain reactions initiated by thermolysis are 1.5 order, first order in the component reacting in the rate-controlling step, and 0.5 order in the initiator. When the initiator is the same component as that reacting in the ratecontrolling step, the reaction will be 1.5 order in this reagent. When chain reactions are initiated by photolysis instead of thermolysis, the rate constant for initiation, the
138 RADICALS
initiator concentration, and the efficiency term F are replaced by the quantum efficiency of the photochemical reaction and either the photon flux, for solutions that absorb all light, or the initiator’s molar extinction coefficient and concentration, for solutions that transmit light.
3.3. Radical Nonchain Reaction Sequences
Radical chain reactions are by far the major class of radical reactions encountered in organic chemistry journal articles and are involved in the bulk of industrial applications of radical chemistry, but radical nonchain sequences are ubiquitous in Nature and arguably provide the largest number of types of radical conversions. In a radical nonchain reaction sequence, the velocity of a termination step is greater than the velocity of at least one potential propagation step. In a perfect nonchain reaction sequence, each initiation reaction would result in a single molecule of product, but that is difficult to achieve in condensed-phase reactions. Nature can accomplish highly efficient radical nonchain reactions by isolating the radical in an enzyme, and there are many such enzyme-catalyzed reactions.
Radical nonchain reaction sequences can result in high yields of a specific pro-
duct when one of the radicals is persistent in solution, and the description of the phenomenon is known as the persistent radical effect.35,36 The model of the radical
chain reaction sequence is changed such that one radical self-termination reaction does not occur or has a slow rate. The radical that does not self-terminate efficiently is the persistent radical, and it accumulates to a relatively high concentration early in the reaction. As the concentration of the persistent radical increases, the velocities of cross-termination reactions involving the persistent radical, which still have diffusion-controlled rate constants, increase such that these are the only important termination reactions. Moreover, the velocity of the cross-termination reaction will eventually exceed the velocity of a propagation step in a normal chain sequence. At this point, the chain sequence is aborted, and the overall process can become a highly efficient nonchain reaction with products formed only from the cross-termi- nation reaction. The negative side of radical nonchain sequences is that a stoichiometric amount of initiator is needed, which can result in high yields of an undesired byproduct. If the initiator is the precursor to the radical that will be functionalized, however, this is not an added problem.
Radical chain processes ‘‘break down’’ whenever the velocity of a termination reaction is comparable to the velocity of the rate-controlling step in a chain reaction. This situation would occur, for example, if one attempted to use Et3SiH as the hydrogen atom donor in the alkyl halide reduction sequence in Figure 4.6 and employed typical ‘‘tin-hydride’’ reaction conditions because the rate constant for reaction of the silane with an alkyl radical is 4 orders of magnitude smaller than that for reaction of Bu3SnH. Such a slow reaction would not lead to a synthetically useful nonchain sequence, however, because no radical is persistent in this case. In fact, a silane-based radical chain reduction of an alkyl halide could be accomplished successfully if the velocity of the initiation reaction was reduced enough such that it (and, hence, also the velocity of alkyl radical termination
MULTISTEP RADICAL REACTIONS |
139 |
reactions) was much less than the velocity of the silane reaction with the alkyl radical. However, this would require inert solvents and an unacceptable increase in the total reaction time.
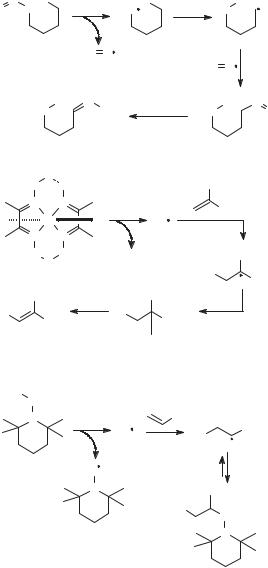
Some examples of radical nonchain reactions are shown in Figure 4.8. In the Barton reaction (first example in Fig. 4.8),37 photolysis of a nitrite ester gives an
O N O H |
|
hν |
O H |
|
O H |
||
Barton Reaction |
O |
N |
|
|
|
||
|
|
|
|
|
|
O N |
|
|
O H |
N OH |
|
O H N O |
|||
|
|
|
|
polar |
|
|
|
|
|
|
|
rearrangement |
|
|
|
O H O |
|
|
|
CN |
|
||
N |
|
N |
|
hν |
|
OEt |
|
pyr |
Co |
|
R |
R |
|
||
|
|
|
|
||||
N |
|
N |
|
|
|
|
|
O |
H |
O |
|
Co(II) |
|
CN |
|
|
|
|
|
R |
|
||
|
|
|
|
|
|
OEt |
|
|
|
|
|
|
|
|
|
CN |
|
|
CN |
|
|
||
R |
OEt |
|
R |
OEt |
|
|
|
|
polar |
Co(II) |
|
||||
|
|
|
|
|
|
|
|
|
|
|
|
Co(III) |
|
|
|
R O |
|
|
|
|
|
|
|
N |
|
|
|
R |
Ph |
|
Ph |
|
|
|
|
R |
|
||
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
O |
|
|
tempo |
|
|
|
|
|
|
|
|
|
|
|
|
N |
|
|
Ph |
|
|
|
|
|
|
R |
O |
|
|
|
|
|
|
|
|
|
|
|
|
tempo |
|
N |
|
|
Figure 4.8. Some radical nonchain reactions.
140 RADICALS
alkoxyl radical and nitric oxide, which is the persistent radical, the alkoxyl radical abstracts hydrogen from a remote carbon, and the carbon radical thus formed reacts with nitric oxide. The reaction of the cobalt complex in the second example is modeled in part after reactions of enzymes that employ coenzyme B12 for radical production; in this case, the Co(II) species formed by photolysis is the persistent radical.38 The third example shows an application of the persistent radical effect in a recently developed polymerization sequence that is one type of living radical polymerization.39,40 Hydroxylamine ethers dissociate thermally to give carbon radicals and nitroxyl radicals, which are persistent. Following a radical addition step, the nitroxyl radical traps the extended polymeric radical. Subsequent dissociation of the new hydroxylamine ether regenerates the polymeric radical that adds to another monomer, and so on. Living radical polymerizations achieve narrow molecular weight distributions because, effectively, no radical termination reactions occur other than the cross-termination reaction. In these examples of radical nonchain reactions, recombination of the persistent radical with the initially formed reactive radical is thermodynamically favored, but such recombination only regenerates the starting materials.
4. ELEMENTARY RADICAL REACTIONS
4.1. Initiations: Radicals from Closed-Shell Compounds
Radicals are formed in thermal and photochemical reactions that homolyze bonds and in electron-transfer processes. In synthetic applications of radicals, a thermal initiator is most often employed to generate radicals. Thermal initiators are also widely used in industrial polymerization applications, but many commercial products employ photoinitiators that can be used to initiate a polymerization after a formulation of monomers is applied. Electron-transfer reactions, either reductions or oxidations, can produce radicals, but strong reducing and oxidizing agents will further reduce or oxidize the radical; therefore, this method requires that the reducing or oxidizing agent reacts relatively slowly with the initially formed radical.
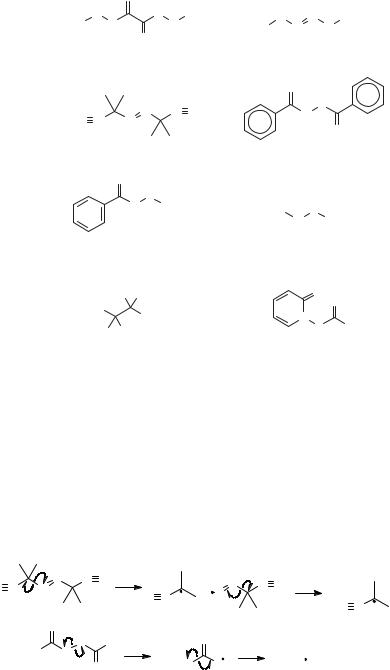
4.1.1. Thermolysis Reactions. Any organic compound will react to give radicals if heated to a high enough temperature, but relatively low-temperature homolysis reactions are possible with azo and peroxy compounds, which contain weak heteroatom–heteroatom bonds.41 Figure 4.9 shows the more common thermal radical initiators. Reactive species such as di-tert-butyl peroxyoxalate42 and di-tert- butyl hyponitrite43 are too unstable to be stored for long periods and are prepared soon before use, but AIBN, diacyl peroxides, peroxy esters, and dialkyl peroxides are commercially available. In synthetic applications of radical chain reactions, it is common to employ a reaction temperature that will result in about a 1-h half-life for the initiator so that low concentrations of radicals will be maintained. For some of the initiators in Figure 4.9, the temperatures that give 1-h half-lives are as follows:44 di-tert-butyl peroxyoxalate, 45 C; di-tert-butyl hyponitrite, 55 C; AIBN, 81 C;
|
|
|
ELEMENTARY RADICAL REACTIONS 141 |
||||
|
|
O |
|
|
|
|
|
O |
O |
O t-Bu |
O |
|
N |
t-Bu |
|
t-Bu |
O |
t-Bu |
N |
|
O |
||
|
|
|
|
|
|||
|
|
O |
|
|
|
|
|
Di-tert-butyl peroxybenzoate |
Di-tert-butyl hyponitrite |
||||||
|
|
|
|
|
O |
|
|
|
|
N |
C N |
|
|
O |
|
N C |
|
|
O |
|
|||
|
N |
|
|
|
|
O |
|
|
|
|
|
|
|
|
|
Azobisisobutyrylnitrile (AIBN) |
Benzoyl peroxide |
||||||
|
|
O |
|
|
|
|
|
|
|
O |
|
|
|
|
|
|
|
O |
t-Bu |
t-Bu |
O |
O |
|
|
|
|
|
|
|||
|
|
|
|
|
t-Bu |
||
|
|
|
|
|
|
||
tert-Butyl peroxybenzoate |
Di-tert-butyl peroxide |
||||||
|
|
Ph Ph |
|
|
|
S |
O |
Bu3SnO |
|
|
|
|
|
|
|
|
OSnBu3 |
|
N |
|
|
||
|
|
|
O |
R |
|||
Ph |
Ph |
|
|
|
|||
|
|
|
|
|
|||
Bis(tributylstannyl)benzopinacolate |
PTOC ester or Barton ester |
||||||
Figure 4.9. Common thermal initiators. The acronym for pyridine-2-thioneoxycarbonyl is PTOC.
benzoyl peroxide, 91 C; tert-butyl peroxybenzoate, 125 C; di-tert-butyl peroxide, 150 C. Many other thermal radical initiators can be prepared; for example, bis- (tributylstannyl)benzopinacolate, prepared from benzopinacol and tributylstannyldimethylamine,45 decomposes at 80 C to give benzophenone and tributylstannyl radicals. Diacylperoxides from alkanoic acids decompose at lower temperatures than does benzoyl peroxide.
The initiation reactions for AIBN and for a diacylperoxide are shown in Figure 4.10. Initial homolysis gives radicals that further react by loss of nitrogen
N C N |
N |
C |
N |
|
|
+ N |
N |
C N |
|
|
|
|
|
|
|
||||
|
|
|
N C |
|
|
2 |
+ N2 |
||
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
N C |
|
|
O |
|
|
|
|
O |
|
|
|
R |
|
O O |
R |
|
|
|
|
|
|
|
2 |
R |
O |
|
2 R + CO2 |
|
|||
|
|
|
O |
|
|
||||
|
|
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
Figure 4.10. Thermal initiation reaction sequences for AIBN and for a diacyl peroxide.