

Reactive Intermediate Chemistry
.pdf244 ORGANIC RADICAL IONS
to the b carbon. However, oxetane (76) is only a minor product, because its radical cation is likely to fragment. Oxetane (77), in which the carbonyl oxygen is connected to the a carbon, is less favorable because one spin of 77 is localized; 77 is built up, however, because 77þ is less likely to undergo fragmentation. The CIDNP results indicate that 76 is formed from a triplet pair, on a time scale comparable with BET in singlet pairs.249
|
|
|
|
|
|
|
Cl4 |
|
|
|
Cl4 |
|
|
|
|
• |
|
H |
|
|
|
|
H O |
|
|
O |
• |
O |
O• |
H |
|
|
|||
H |
|
• |
|
|
||||
|
|
|
|
|
|
|
|
|
|
76 • • |
|
77 • • |
|
|
|||
When carried out in the presence of molecular oxygen, PET reactions between some donor–acceptor pairs yield oxygenated products. The radical anions of accep-
tors with appropriate reduction potentials reduce O2 to superoxide ion, O2 , which then couples to the cations.250–252 In the PET reaction between trans-stilbene and
9-cyano-phenanthrene, the trans-stilbene radical cation was observed by optical
spectroscopy and the 9-cyanophenanthrene radical anion by ESR; the anion spectrum decayed rapidly in the presence of oxygen.251,253 Other details of the mechan-
ism, for example, the spin multiplicity of the pair or the detailed mechanism of adduct formation, are not known.
The type of O2 adduct depends on the donor structure. For example, tetraphenyloxirane forms an ozonide (78),254 1,4-bifunctional radical cations form dioxanes
(79);255 conjugated dienes form cyclic adducts (80);256 and ergosteryl acetate (81) forms the 5a,8a-peroxide (82) at 78 C.257
Ph |
O |
Ph |
Ph |
|
Ph |
|
|
|
|
|
|
|
|
||||||
|
|
|
|
|
|
||||
Ph |
|
O |
Ph |
Ph |
|
Ph |
O O |
||
O |
|
|
|
O O |
|||||
|
78 |
|
|
79 |
80 |
|
|||
|
|
|
|
C9H17 |
|
|
|
|
C9H17 |
AcO |
|
|
|
|
|
AcO |
O O |
||
|
|
|
|
|
|||||
|
|
|
|
|
|||||
|
|
|
|
|
|||||
81 |
|
|
|
|
|
82 |
|
||
There are, however, additional mechanisms for the oxygenation of radical cations, which do not qualify as intra-pair reactions. For example, hindered, a-branched tetraalkylalkenes (e.g., biadamantylidene) form dioxetanes via a chain

RADICAL CATION REACTIONS: RELATIONSHIPS WITH OTHER INTERMEDIATES |
245 |
mechanism, initiated by chemical, electrochemical, or PET oxidation.107,258,259 The hindered radical cation reacts with triplet oxygen, forming a bifunctional adduct that undergoes ring closure to a dioxetane radical cation of type 5þ, with a three-electron bond (see above); ET from the alkene generates 5; this step is efficient, because 5þ (Eox ¼ 2:3 V vs. SCE, 78 C) is a better oxidant than the alkene radical cation (Eox ¼ 1:6 V vs. SCE, 78 C).
|
O • • |
• O |
|
|
+ • |
|
|
|
||
+ • |
O |
|
O O |
|
|
O O |
||||
2 |
|
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
–  +•
+•
5 • +
In some cases, the structures of oxygenation products have been crucial for assigning the structures of unusual radical cations. For example, the endo-peroxides (83 and 85) support the structures assigned to radical cations (24þ and 84þ)
derived from 1,1-diaryl-2-methylenecyclopropane (23) and 2,5-diaryl-1,5-hexadiene, respectively.241,255 Time-resolved spectroscopic data suggest that 83 is generated
by coupling of triplet biradical (24 ) with (triplet) molecular oxygen.241
|
|
|
|
• * |
|
Ar |
||
|
# |
|
|
|
Ar |
|||
• |
Ar |
|
O2 |
|
|
|||
|
Ar |
|
|
|
|
|
O |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
O |
|
24 • # ; # = +, • * = – , • |
83 |
||||||
|
|
|
• + |
|
O |
|||
|
|
|
Ar |
|
Ar |
|||
|
|
|
|
O |
||||
|
|
|
|
|
O2 |
• – |
||
Ar |
|
|
|
|
|
|
|
Ar |
|
|
|
|
|
|
|
|
|
|
84 • + |
|
|
|
|
85 |
||
Ar = aryl
Radical ion pairs of triplet multiplicity can couple via charge recombination, equivalent to nucleophilic capture of the radical cation by the radical anion, yielding biradicals. In reactions of this type, radical cations with stereochemically distinct faces offer a choice of stereochemical approach; the isomers 15þ and 16þ
provide illuminating examples. These species react with methanol in stereospecific fashion from the exo-face,260,261 forming exo-methoxy-norbornyl and -nortricyclyl
free radicals, which undergo rapid cyclopropylcarbinyl–butenyl interconversions. Likewise, benzoquinone radical anion approaches 15þ and 16þ stereoselectively from the exo-face.262 The resulting biradicals (86 and 87 ) undergo cyclopropyl- carbinyl–butenyl interconversions, equilibrating 86 and 87 with 88 . The ‘‘preoxetane’’ (86 ) and the ‘‘pre-oxolane’’ (88 ) form products 86 and 88,

246 ORGANIC RADICAL IONS
respectively, after intersystem crossing. One of the primary biradicals (87 ) cannot cyclize, because the unpaired spins are mutually inaccessible.
|
|
|
O • |
|
|
|
|
|
O |
O |
|
• |
O |
O • |
• |
|
|
|
• |
|
|
|
|
|
86 • • |
O • |
|
87 • • |
88 • • |
O
O
 O
O
O
86 |
88 |
5.3. Bimolecular Radical Ion Reactions
The majority of radical ion reactions are bimolecular in nature, although some of these are merely variations of the unimolecular reactions discussed above, and many occur as pair reactions, albeit with a modified ‘‘partner.’’ Radical ions may react with polar or nonpolar neutral molecules, with ions, with radicals, or with radical ions of like or opposite charge (Scheme 6.3). Alkene radical ions undergo a particularly rich variety of reactions, including additions and cycloadditions.
5.3.1. Reactions with Alkenes and Aromatics. Unsymmetrically substituted
olefins form head-to-head dimers selectively. We mention the ‘‘classic’’ vinylcarbazole,41 vinyl ethers,157,263,264 indenes,265–267 and p-methoxystyrene.268 The regio-
chemistry of the addition is compatible with a stepwise mechanism proceeding via a singly linked 1,4-bifunctional radical cation, in which spin and charge are
separated (see above). Several dimerizations have quantum yields greater than unity;157,263,269 these results support radical cation chain processes.
1,1-Diphenylethylene radical cation forms a dimeric product (90) via a 1,4- bifunctional–distonic intermediate (89þ) that undergoes an alternative (1,6-) cyclization with participation of one phenyl group.270
|
Ph |
|
Ph |
• + |
Ph |
||
|
|
|
|
|
|||
Ph |
+ |
|
|
|
|
|
|
Ph |
• |
|
H |
|
|
|
Ph Ph |
|
|
|
|
||||
|
|
|
|
||||
|
|
|
|
|
|
||
|
Ph |
|
Ph Ph |
|
|
|
|
|
89 • + |
|
|
|
|
|
90 |

RADICAL CATION REACTIONS: RELATIONSHIPS WITH OTHER INTERMEDIATES |
247 |
When generated in zeolites, alkene or arene radical cations react with the parent molecules to form p-dimer radical cations. For example, 2,3-dimethyl-1-butene271 and benzene272 formed 91þ and 92þ, respectively. The confinement and limited diffusion of the radical cation in the zeolite favor an interaction between a radical cation and a neutral parent in the same channel.
• + |
|
|
• + |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
91 • + 92 • +
Similarly, fluorescence detected magnetic resonance effects observed during the pulse radiolysis of anthracene-d10 in the presence of 2,3-dimethyl-1-butene support the presence of 8 equivalent methyl groups. Because the splitting, adi ¼ 0:82 mT, was approximately one-half that of the monomer splitting, amon ¼ 1:71 mT, the ‘‘sandwich’’ dimer 91þ was invoked.273
However, ab initio calculations [QCISD-(T)/6-31G*//UMP2/6-31G*] on ethylene and its radical cation support an ‘‘anti’’-p-complex, in which the two components are joined by one long bond (190 pm), rather than the ‘‘sandwich’’-type p complex. The complex is connected to two transition states leading to a (rhombic) cyclobutane radical cation (see above) or, by 1,3-H-shift, to 1-butene radical cation.274
|
• |
+ |
|
|
|
|
|
|
|
• + |
|
• + |
|
|
|
|
CH |
|
H2C |
|
CH2 |
|
|
||||
|
|
|
|
|
|
||||||||
H2C |
|
|
|
||||||||||
|
|
|
|
|
|
|
|
|
|||||
|
|
|
H2C |
|
CH3 |
|
|
|
H2C |
|
CH2 |
|
|
|
|
|
|
|
|
|
|
|
|
||||
Irradiation of naphthalene in faujasite pores gave rise to loading-dependent nanosecond time-resolved diffuse reflectance spectra.275 A structured band (680 nm) at low loadings was assigned to the monomer radical cation, whereas a broad band (590 nm) at higher loading was assigned to the dimer radical cation. The dimer structure is supported by a broad charge resonance band in the nearinfrared (IR) ( 1100 nm); it was assigned a ‘‘twisted’’ structure with partial overlap of the two p systems.
Some radical cations with spin and charge on heteroatoms form s-dimer radical cations. For example, irradiation of acetophenone and aliphatic amines inside Na Y zeolite generated amine dimer radical cations (93þ; lmax 480–580 nm).108 Analogous s-dimer radical cations are derived from thiols.276
O |
|
|
|
hν |
|
|
|
|
|
|
|
|
|
|
R |
||
|
|
|
|
|
|
• + |
|
|
|
|
|
|
R |
• + |
|||
|
Me |
|
|
|
|
|
|
|
|
|
|
|
|
R |
|||
|
|
|
|
• • |
|
|
|
|
|
• • |
|
||||||
|
|
|
|
|
N |
|
|
|
|
|
N |
N |
|||||
|
|
R |
|
N |
R |
R |
R |
|
N |
R |
|||||||
|
|
|
R |
|
|
|
|||||||||||
|
|
|
R |
|
R |
|
R |
||||||||||
|
|
|
|
R |
|
|
|
|
R |
R |
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
93 • + |
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||

248 ORGANIC RADICAL IONS
Radical cations (94þ) of simple alkynes (e.g., butyne) generated by g-irradia- tion in frozen matrices are stable at 77 K. Upon warming, changes in the ESR
spectra support an interesting cycloaddition forming cyclobutadiene radical cations (95þ).277
• + |
|
|
R' |
• + |
R' |
|
|
R' |
|
|
|
|
||
C |
|
|
|
|
C |
|
|
|
|
|
|
|
|
|
R |
|
R |
|
R |
|
|
|
|
|
94 • + |
|
|
|
95 • + |
PET converts phenylacetylene to a-phenylnaphthalene; a 1,4-bifunctional dimer radical cation (96þ) is the key intermediate; 1,6-cyclization, followed by a hydrogen or hydride shift generates the final product.278
Ph |
• + |
|
|
• + |
• + |
|
|
|
|
|
|
|
|
C |
|
|
|
|
|
|
HC |
|
|
|
|
|
|
|
|
|
|
|
|
|
HC |
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
Ph |
H |
||
Ph |
|
|
Ph |
|||
|
|
|
|
|
|
|
|
|
|
96 • + |
|
||
PET reactions of diolefins (viz., cyclohexadiene) generate [4 þ 2] cycloadducts (e.g., endoand exo-dicyclohexadiene).279 This conversion occurs also upon radiolysis280 or chemical oxidation [tris( p-bromophenyl)aminium salts; cation radical catalyzed Diels–Alder reaction]. The scope of this reaction and its synthetic utility have been delineated in detail; once again, a radical cation chain mechanism is indicated.281
|
H |
hν |
H |
|
+ |
ET sens |
H |
|
|
|
H |
The mechanism of the cycloaddition appears to be concerted for various reagents; however, for several cases, radical cation cycloaddition–cycloreversions have a stepwise component. For example, CIDNP effects observed during the PET induced dimerization of spiro[2.4]heptadiene (97) identify a dimer radical
cation with spin density only on two carbons of the dienophile fragment; this intermediate must be a doubly linked radical cation (99þ).282,283 A pulsed laser experi-
ment at high concentrations of 97 supports a second dimer radical cation; at high

RADICAL CATION REACTIONS: RELATIONSHIPS WITH OTHER INTERMEDIATES |
249 |
concentrations of 97, the dimer cation is rapidly quenched by ET from the
monomer. This rapid quenching allows the polarization of a second, singly linked dimer radical cation (98þ) to be observed.282,283
 •
•
|
|
+ |
+ |
|
|
• |
|
|
|
|
|
97 |
98 • + |
|
99 • + |
In some cases, a stepwise mechanism is indicated by randomization of the dienophile stereochemistry. For example, addition of cis-anethole radical cation (100þ) to cyclopentadiene produces comparable yields of four possible diastereoisomeric adducts (102) clearly supporting a distonic radical cation intermediate
(101þ). Only products supporting the stepwise mechanim, that is, trans,endoand trans,exo-102, are shown.284
An |
H |
|
|
|
An |
|
|
|
|
|
|
||
|
|
+ |
|
|
|
|
+ |
|
An |
|
|
Me |
|
|
|
• |
|
|
|
+ |
• |
|
|
|
|
||
|
|
|
|
An |
||
Me |
|
|
|
|
||
Me |
|
|
Me |
|
||
|
|
|
|
|
||
|
|
|
|
|
|
|
100 • + |
101 • + |
|
|
trans,endo-102 |
trans,exo-102 |
|
An = C6H4OMe |
|
|
|
|
|
|
In the ET-catalyzed Diels–Alder reaction of indole with 1,3-cyclohexadiene a stepwise mechanism was derived on the basis of 13C kinetic isotope effects. In light of B3LYP/6-31G* calculations, these effects support a stepwise mechanism, initiated by attack of the diene on the 3-position of indole.285
Of the many PET Diels–Alder reactions of potential synthetic utility we mention two reactions of vinylindole (103) catalyzed by 2,4,6-tris(4-methoxyphenyl)- pyrylium tetrafluoroborate. With cyclohexadiene, 103 reacts as a diene, giving rise to tetrahydrocarbazole (104);286 with exocyclic dienes, 103 serves as dienophile generating a different tetrahydrocarbazole (105).287 Molecular orbital calculations provide a rationale for the regioand diastereoselectivities of these reactions.

250 ORGANIC RADICAL IONS
N  Me
Me
H
CN
104
Me
N |
Me |
|
N |
|
|
|
|||
H |
CN |
|
H |
Me |
|
|
|||
|
|
NC |
||
|
103 |
|
Me |
|
|
|
|
|
|
|
|
|
|
Me |
|
|
|
|
105 |
5.3.2. Reactions with Protic, Ionic, Polar Reagents. The reactions of radical anions with proton donors include the reduction of arenes,288 the well-known Birch reduction, as well as alkynes289 by alkali metals in liquid ammonia. Both reactions have synthetic utility and belong to the few radical ion reactions included in elementary textbooks.
|
|
|
|
|
|
|
|
|
|
|
H |
H |
|
• |
|
|
H+ |
|
• |
|
e– |
|
|
|
|
|
|
|
|
|
|
|||||||
|
– |
|
|
|
|
|
|
H+ |
|
|
|
|
|
|
|
|
|
H H |
|
H |
H |
||||
|
|
|
|
|
|
|
|
|
||||
106 • – |
|
|
|
107 • |
|
|
|
|
|
|||
Following the reduction of the substrates by a solvated electron, the solvent transfers a proton to the radical anions, 106 and 108 ; the resulting radicals, 107 and 109 , are then reduced again, and the anions, 107 and 109 , are protonated once more. The regiochemistry of the protonation of 107 is kinetically controlled; the ready inversion of the alkenyl free radical 109 is the key to the formation of the trans-alkene.
R |
• – |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
|
|
* |
|
|
R |
|
|
|
R |
* |
|
|
|
|
|
H |
||||
|
|
|
|
|
|
|
|
|
|
|
R |
|
|
|
||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||||||
C |
|
H+ |
C |
|
|
|
|
|
C |
e– |
C |
|||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
C |
|
|
C |
R |
|
|
|
|
C |
H+ |
C |
|||||||||
|
|
H |
|
|
|
H |
||||||||||||||
R |
|
|
|
|
|
|
R |
|
H |
|
|
|
R |
|||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
108 • – |
|
|
|
|
|
109 * ; * = • , – |
|
|
|
|
|
|
|
|
||||||

RADICAL CATION REACTIONS: RELATIONSHIPS WITH OTHER INTERMEDIATES |
251 |
With ions or dipolar substrates, radical ions undergo nucleophilic or electrophilic capture. Nucleophilic capture is a general reaction for many alkene and strainedring radical cations and may completely suppress (unimolecular) rearrangements or dimer formation. The regioand stereochemistry of these additions are of major interest. The experimental evidence supports several guiding principles.
For example, the PET reaction of 1-phenylcyclohexene (110) in the presence
of nucleophiles (KCN; acetonitrile / 2,2,2-trifluoroethanol) proceeds by antiMarkovnikov addition of the nucleophile to the alkene.270,290 A distonic oxonium
radical cation, 111þ, is a likely short-lived intermediate, which is rapidly deprotonated.
Ph |
KCN |
+ |
Ph |
R–OH |
Ph |
|
H |
|
H |
||||
|
• |
|
|
|
||
|
|
|
|
|||
|
|
|
|
|
|
|
CN |
|
|
H |
|
|
OR |
|
|
110 |
• + |
|
|
|
Ph
•
H
R
O + H
111 • +
The nucleophilic capture of radical cations forms (free) radicals, one H atom shy of the adduct. The missing H may be introduced in one step, by hydrogen abstraction, or in two, involving successive reduction by the sensitizer radical anion and protonation. Both mechanisms have been observed, sometimes in competition with each other.
Substituted cyclopropane systems also undergo nucleophilic addition of suitable solvents (MeOH).291 For example, the photoinduced ET reaction of 1,2-dimethyl-3- phenylcyclopropane (112, R ¼ Me) with p-dicyanobenzene formed a ring-opened ether by anti-Markovnikov addition. The reaction occurs with essentially complete inversion of configuration at carbon, suggesting a nucleophilic cleavage of a ‘‘oneelectron’’ cyclopropane bond, generating 113 .292 The retention of chirality confirms that the stereochemistry of the parent molecule is unperturbed in the radical cation 112þ.
Ph |
|
|
H |
|
R |
|
R |
• |
|
ET |
Ph |
|||
|
||||
H H |
|
|
||
MeOH |
|
H |
||
|
|
|
||
R |
|
MeO |
R |
|
112 |
|
|
113 • |
Many of these reactions support a measure of ‘‘thermodynamic control’’ in nucleophilic capture: Conjugated radicals or products formed with release of ring strain are favored. For example, the addition of ethanol to radical cation 110þ is regiospecific, forming the more stable (benzylic) intermediate 111þ; the capture of 112þ likewise forms a benzylic radical (113 ). Radical cation 48þ generates a
252 ORGANIC RADICAL IONS
conjugated radical cation 49þ via a sigmatropic shift, and an allyl radical 114 upon nucleophilic attack;211 both reactions form conjugated ‘‘products’’ with relief of ring strain. The high degree of regioselectivity in the nucleophilic capture of 48þ, 110þ, or 112þ also reflects unfavorable energetics for the formation of alternative products.
• + |
|
|
• + |
||
|
|
|
|
|
• |
|
|
|
|
|
OMe |
|
|
|
|
|
|
49 • + |
|
48 • + |
|
|
114 • |
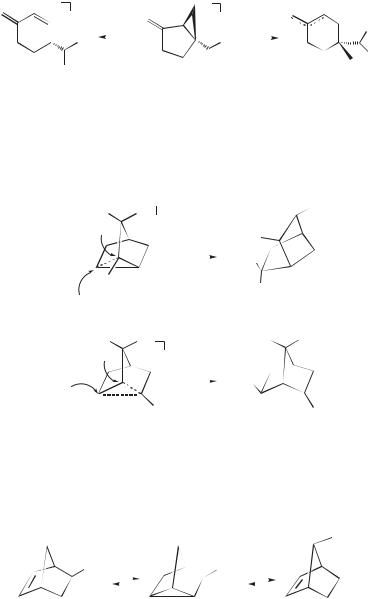
The nucleophilic capture of tricyclane radical cations 115þ and 117þ supports
the role of ‘‘conventional’’ steric hindrance; 115þ reacts at the 3 carbon (!116 ),293 whereas the chiral isomer 117þ is captured by backside attack at
the less hindered 3 carbon (!118 ).294 Both reactions are regioand stereospecific and avoid attack at the neopentyl-type carbon (denoted by an asterisk).
|
|
• + |
||
|
Nu |
|
|
• |
|
|
|
|
|
|
* |
|
|
H |
|
|
|
||
|
Nu |
|
|
MeO |
|
|
|
|
|
|
115 • + |
|
|
116 • |
|
|
• + |
||
|
Nu |
|
|
|
Nu |
* |
|
|
MeO |
|
|
|||
|
|
|
|
• |
|
117 • + |
|
|
118 • |
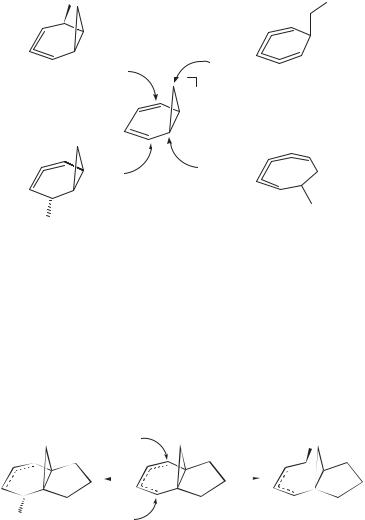
The radical cations 15þ and 16þ add methanol exclusively from the exo face forming exo-methoxynorbornyl and nortricyclyl free radicals, which undergo
rapid cyclopropylcarbinyl–butenyl interconversions (119 Ð 120 Ð 121 ; see above).260,261 The attack on 15þ can be viewed as a backside attack of the nucleo-
phile (Nu) on the weakened cyclopropane bond with inversion of configuration.
|
|
|
|
|
|
|
|
|
|
OMe |
|
OMe |
OMe |
||||||||
• |
|
|
|
|
• |
|
|
|
|
• |
|
|
|
||||||||
|
|
|
|
|
|
|||||
|
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
119 • |
120 • |
121 • |
RADICAL CATION REACTIONS: RELATIONSHIPS WITH OTHER INTERMEDIATES |
253 |
|
OMe |
OMe |
|
. |
. |
|
∆G = 0.4 kcal/mol |
• + |
|
∆G = –18.7 kcal/mol |
||
|
. |
. |
|
|
|
OMe |
MeO |
|
∆G = 0.0 |
∆G = –13.9 kcal/mol |
Scheme 6.8
However, orbital factors may override thermodynamic control. For example, the regiochemistry of nucleophilic attack on the bridged norcaradiene radical cation 122þ shows a significant deviation from thermodynamic control. Although attack on the cyclopropane ring should be favored by both release of ring strain and formation of delocalized free radicals (cf. Scheme 6.8), methanol attacks 122þ selectively at C2 (and C5), generating 123 and 124 . There is little stereoselectivity: Products derived from 123 and 124 were formed in comparable yields.152
|
|
|
|
OMe |
• |
|
• |
|
. |
MeO |
|
+ |
|
|
|
122 • + |
|
|
|
123 • |
|
|
124 • |
The bridged norcaradiene (122þ) appears particularly well suited to evaluate orbital and thermodynamic effects in radical cation reactions because these effects predict different reactivity patterns and because the orbital coefficients of SOMO and LUMO also differ significantly (Fig. 6.16). The two MOs potentially involved in the reaction have a substantially different distribution of orbital coefficients. The SOMOs of 122þ have large orbital coefficients at C2,5 and C7 (C10), which are reflected in the hyperfine pattern of these species. In contrast, the principal orbital coefficients of the LUMOs are located at C2,5 and C3,4 and the orbital lobes at C7 (C10) offer no target for an attack by the nucleophile. Because the orbital coefficients of SOMO and LUMO differ, the norcaradiene system will elucidate whether