

Reactive Intermediate Chemistry
.pdf122 RADICALS
1. STRUCTURE AND REACTIVITY
1.1. Structure
Organic radicals are species that contain an unpaired electron in the highest energy (singly) occupied molecular orbital, referred to as SOMO for semioccupied MOs. The structures of organic radicals are familiar to most chemists, who were introduced to radicals in their first organic chemistry course. As with closed-shell molecules, conformational interconversions of radicals are low-energy processes. Unlike closed-shell molecules, however, configurational interconversions of radicals are also often low-energy processes. Radical configurations are described according to whether the SOMO is a p-type orbital (p radical) or a hybrid orbital (s radical). Examples of organic radicals are shown in Figure 4.1.
A trivalent p radical is planar, and a trivalent s radical is pyramidyl. Both types of trivalent radicals have staggered and eclipsed conformations. For a planar radical, staggered and eclipsed refer to the substituents at the radical center and not to the p orbital. A divalent p radical is linear, and a divalent s radical is bent. In the case of heteroatom radicals, such as alkoxyl and aminyl radicals, two low-energy electronic states exist, and the odd electron can be in a p orbital with the lone pair in a hybrid orbital (p radical) or vice versa (s radical).
Interconversions of acyclic carbon-centered radicals between p and s types are low-energy processes. The methyl radical is planar, but increasing alkyl substitution at the radical center results in an increasing preference for pyramidalization. The tert-butyl radical is pyramidalized with the methyl groups 10 from planarity (the deviation from planarity for a tetrahedral atom is 19 ) and a barrier to inversion of 0.5 kcal/mol.1 When a radical center is in a carbocycle, a planar radical is favored for all cases except the cyclopropyl radical, and the barrier for inversion in cyclopropyl is only 3 kcal/mol.2
Substitution of electronegative groups for hydrogen in the methyl radical favors pyramidalization, and the trifluoromethyl radical is tetrahedral.3 This effect is a result of interaction of the SOMO with the lowest unoccupied molecular orbital (LUMO). The SOMO in the methyl radical is a carbon 2p orbital, and it is orthogonal to the LUMO, whereas, in a pyramidal radical, the SOMO is a hybrid orbital that can interact with the LUMO. Electronegative groups increase the energy of the SOMO by p donation and decrease the energy of the LUMO by s withdrawal. As the energy levels of the SOMO and the LUMO approach one another, interaction
H
H |
|
H |
|
H |
F |
|
H |
H |
|
F |
|
||
|
|
F |
H |
|||
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
(a) |
(b) |
(c) |
|
(d ) |
Figure 4.1. The (a) ethyl (shown in a staggered conformation) and (b) cyclohexyl radical are p radicals, and the (c) trifluoromethyl radical and (d) cyclopropyl radical are s radicals.
STRUCTURE AND REACTIVITY |
123 |
between them is increasingly favored, and a s radical becomes the low-energy configuration. On the other hand, conjugation of the trivalent radical center with a p system favors a planar structure. For example, in the case of the benzyl radical, the energy required for deformation from the planar structure in the transition states for reactions results in rates of additions to alkenes that are slower than expected from thermodynamic considerations.4
Divalent radicals are usually s radicals. The vinyl radical is bent, and the barrier for inversion through the linear form is 3 kcal/mol.5 Vinyl radicals with sigma substituents also are bent, but p substituents give linear vinyl radicals.6 The formyl radical and acyl radicals are bent.7
The conformational barriers in acyclic radicals are smaller than those in closedshell acycles, with the barrier to rotation in the ethyl radical on the order of tenths of a kilocalorie per mole.1,8 The barriers increase for heteroatom-substituted radicals, such as the hydroxymethyl radical, which has a rotational barrier of 5 kcal/mol. Radicals that are conjugated with a p system, such as allyl, benzyl, and radicals adjacent to a carbonyl group, have barriers to rotation on the order of 10 kcal/mol. Such barriers can lead to rotational rate constants that are smaller than the rate constants of competing radical reactions, as was demonstrated with a-amide radicals,9 and this type of effect permits acyclic stereocontrol in some cases.10
1.2. Radical Stabilities and C H Bond Dissociation Energies
Radical stabilization energies (RSEs) and bond dissociation energies (BDEs) are critically important values for understanding radical reactivities. Homolytic BDEs are strongly related to the stabilities of the radicals formed by homolysis reactions, although BDEs and RSEs are not exactly equivalent.11 Among the most precisely measured BDE values are those for cleavage of bonds to hydrogen
atoms, and C H BDEs for small organic compounds are determined with precisions of 0.5–2 kcal/mol. Several compilations of BDEs are available,12–15 and these
works summarize the methods used to obtain the thermochemical data. When using BDE values, one should ensure that the scales from different sources are equivalent by referring to the BDEs of simple molecules such as methane and water. Table 4.1 contains representative BDE values for small compounds.
The X H BDEs increase across a row of the periodic table, reflecting increasing nuclear charge on atom X, and decrease as one moves down a column in the periodic table. The nuclear charge effect in the second row is dramatic with BDEs increasing by 30 kcal/mol from methane to HF, following the increase in the electronegativity of atom X. The same pattern is observed for hybridization of carbon with BDEs increasing as the amount of s character in the C H bond increases. For example, the BDEs for ethane, ethylene, and acetylene are 101, 111, and 133 kcal/mol, respectively, and the high degree of s character in the C H bonds of cyclopropane results in a C H BDE that is greater than that of methane.
Any substituent on a trivalent C-centered radical is stabilizing. This phenomenon is a result of interactions resulting from mixing the SOMO containing the odd

124 RADICALS
TABLE 4.1. Bond Dissociation Energies (in kcal/mol)a
H2 |
104.2 |
|
HF |
136.4 |
0.2 |
HCl |
103.15 |
0.03 |
HBr |
87.54 |
0.05 |
HI |
71.32 |
0.06 |
H2O |
119.30 |
0.05 |
CH3O H |
104.2 |
0.9 |
H2S |
91.2 |
0.7 |
CH3S H |
87.4 |
0.5 |
NH3 |
108.2 |
0.3 |
CH4 |
104.9 |
0.1 |
CH3CH2 H |
101.1 |
0.4 |
(CH3)2CH H |
98.6 |
0.4 |
(CH3)3C H |
96.5 |
0.4 |
c-C3H5 H |
106 |
|
|
H2CCH H |
111.2 0.8 |
||
Ph H |
111.2 0.8 |
||
HCC H |
132.8 0.7 |
||
CH2CHCH2 H |
88.2 |
|
2.1 |
PhCH2 H |
88.5 |
|
1.5 |
HOCH2 H |
96.06 |
|
0.15 |
NCCH2 H |
94.8 |
|
2.1 |
HC(O)CH2 H |
94.3 |
|
2.2 |
RC(O)O H |
105 |
|
|
CH3C(O) H |
89.4 |
|
0.3 |
HOC(O) H |
>89.5 |
||
[(CH3)3Si]3Si H |
84 |
|
|
Bu3Sn H |
79 |
|
|
a Enthalpies for bond homolysis at 25 C. Data taken from (12–15).
electron with either filled or empty MOs. In all cases, p-type interactions give new MOs of lower and higher energies. When the SOMO mixes with a filled MO, such as the C H s-bonding orbital of the methyl group in the ethyl radical or the lone pair on an adjacent oxygen atom, two electrons occupy the lower energy orbital and one occupies the higher energy orbital. When the SOMO mixes with an empty orbital, such as the LUMO of an electron-withdrawing group, the odd electron resides in the new low-energy combination MO.
Delocalization of the odd electron into extended p systems results in considerable radical stabilization. The C H BDE at C3 of propene is reduced by 13 kcal/mol relative to that of ethane. That the stabilization effect in the allyl radical is due primarily to delocalization in the p system is shown by the fact that the rotational barrier for allyl is 9 kcal/mol greater than that for ethyl.16 Extending the conjugated system has a nearly additive effect, and the C H BDE at C3 of 1,4-pentadiene is 10 kcal/mol smaller than that of propene.12 Delocalization of the odd electron in the benzyl radical results in about one-half of the electron density residing at the benzylic carbon, and the C H BDE of the methyl group in toluene is the same as that in propene.
Computational determinations of radical stabilities and BDE values have advanced to the point that computed and experimental values are in very good- to-excellent agreement, and it is noteworthy that relatively economical hybrid density functional theory computations can give excellent BDE results.17 An advantage of computations is that radical stabilities can be determined for cases that would be difficult to measure; for example, the BDE value for the O H bond in mercaptoethanol can be computed even though the S H bond is weaker. Reliable estimations of BDE values for large molecules that contain isolated groups can be made from the BDE values for small molecules, but computations are recommended for large molecules when functional groups interact with the radical center.
STRUCTURE AND REACTIVITY |
125 |
In addition, computed BDE values are valuable for bonds that do not involve a hydrogen atom because experimental BDE values are mainly for X H and X C bonds.
1.3. Stable and Persistent Radicals
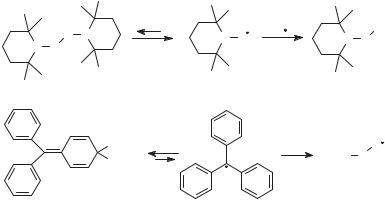
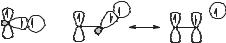
Most radicals are highly reactive and short lived, often reacting with one another with diffusion-controlled rates. Examples of thermodynamically stable radicals are known, however (Fig. 4.2). Nitroxyl radicals lacking b-hydrogen atoms, such as 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), or with bridgehead b-hydrogen atoms equilibrate with their dimers. The nitroxyl radicals are favored at equilibrium because the O O bond formed by coupling is weak. The triphenylmethyl (or trityl) radical also equilibrates with its dimer, a quinoid species, with the dimer favored at equilibrium. An adequate concentration of trityl exists in solution at room temperature such that the solution has a yellow color. This color was observed by Gomberg18 upon reaction of triphenylmethyl bromide with metals and reported in 1900 as evidence for formation of the trityl radical; Gomberg’s work is often considered to be the beginning of organic radical chemistry. In the case of trityl, delocalization of the odd electron in the radical and strain in the dimer are the origins of the stability. Some radicals are thermodynamically disfavored with respect to dimerization but have relatively long lifetimes due to steric hindrance that slows radical–radical reactions.
Although persistent radicals can be thermodynamically favored with respect to their dimers, they often react rapidly with other molecules and radicals. For example, TEMPO couples with alkyl radicals with rate constants that are nearly as large as diffusional rate constants19 to give oxime ethers that are stable
|
O N |
TEMPO |
|
R |
|
|
R |
|
|
N O |
|
|
|||
N O |
|
|
|
N O |
|||
|
|
|
|
||||
|
|
|
|
|
|
|
|
|
|
|
TEMPO |
|
|
Oxime ether |
|
|
|
CPh3 trityl |
|
|
O2 |
|
O |
|
|
H |
|
|
|
Ph3C |
O |
Trityl radical
Figure 4.2. The persistent radicals TEMPO and trityl react rapidly with alkyl radicals and oxygen.
126 RADICALS
at room temperature, and Gomberg noted that the yellow solutions containing trityl lost their color when oxygen was admitted to the reaction mixture. This property can be exploited in synthesis and polymerization chemistry in radical nonchain reactions as discussed in Section 3.3, and it is a feature of many constructive enzyme-catalyzed radical reactions in Nature.
2. IDENTIFICATION AND CHARACTERIZATION OF RADICALS
Despite short lifetimes, radicals can be observed directly by several spectroscopic techniques. A transient radical also can be implicated by the products of its reactions, either closed-shell molecules formed in a specific sequence of radical reactions or ‘‘stable’’ radicals formed by reaction of a short-lived radical with a closed-shell molecule. Several characterization methods for radicals are unique, and they are discussed briefly in this section.
2.1. Inference from Products
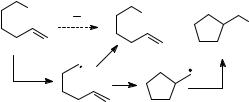
As with any intermediate, a transient radical can be implicated from products formed in a reaction specific to the radical of interest. Experimentally, this is the basis of so-called mechanistic probe studies. An application of this method might employ, for example, 6-bromo-1-hexene as a probe for a radical intermediate as shown in Figure 4.3. If the 5-hexenyl radical is formed as a transient with an adequate lifetime, then cyclization of this radical to the cyclopentylmethyl radical could eventually give the cyclic product, and detection of the cyclic product provides evidence that a radical was formed. The mechanistic probe approach is deceptively simple, however. To be useful, one must exclude other possibilities for formation of the rearranged product and demonstrate that the transient was formed in the reaction of interest and not in a side reaction. The latter is especially difficult to demonstrate, and, unfortunately, some mechanistic probe studies that seemingly provided ‘‘proof’’ of radical intermediates were later found to be complicated by radical-forming side reactions.
Somewhat more definitive evidence that a transient radical was produced is provided when the radical reacts with a trapping agent to give a stable radical product
Br |
X Y |
X |
|
|
X |
||
|
|
+ |
Figure 4.3. Design of a radical probe mechanistic study. Formation of the rearranged product implicates the intermediate 5-hexenyl radical that cyclized to cyclopentylmethyl.
IDENTIFICATION AND CHARACTERIZATION OF RADICALS |
127 |
because these reactions are specific for radicals. Examples include additions of radicals to nitroso compounds or nitrones, both of which give N-oxyl radical products that can be detected by electron spin resonance (ESR) spectroscopy. The technique is known as spin trapping, and can provide structural information about the initial radical that is trapped and quantitative information about the concentrations of radicals formed in addition to the qualitative confirmation that a radical was present.20 One common spin trap is C-phenyl-N-tert-butylnitrone (PBN), which reacts as shown. Nitrones react with oxygen-centered radicals also, and watersoluble nitrones have been used to quantitate oxygen radicals in biological systems, so-called ‘‘active oxygen’’ intermediates, by quantitative ESR methods.21
Ph |
O |
Ph |
O |
|
R |
N |
|||
R |
N |
|||
H |
CMe3 |
H |
CMe3 |
|
|
PBN |
A stable N-oxyl radical |
||
2.2. Indirect Kinetic Determinations
The kinetics of radical reactions can be studied by direct methods (discussed in Chapter 18 of this volume) or by indirect methods. Indirect kinetic studies22 require no special instrumentation, and are popular for obtaining relative or absolute rate constants for an intermediate that might be formed in a specific conversion. The radical of interest is generated in a reaction where two pathways compete, the reaction of interest and the basis reaction, for which a rate constant is known. Relative rate constants are then determined from the product mixture by spectroscopy or chromatography and used with the basis rate constant to calculate the absolute rate constant for the reaction of interest. The method is most easily applied when the competing reactions are first order or pseudo-first order, but that is not a requirement.22
When the basis reaction in the competition kinetic scheme is a calibrated first-
order rearrangement, a cyclization, ring opening, or rearrangement reaction, then the radical is called a radical clock.22–24 Calibrated alkyl radical clocks that cover a kinetic range >12 orders of magnitude are available.22,24 In the example in
Figure 4.3, the 5-hexenyl radical cyclizes with a known rate constant and can serve as a radical clock. The product ratio from the competition reaction is given by Eq. 1, where kXY is the second-order rate constant for reaction of X Y, kC is the first-order rate constant for cyclization of the 5-hexenyl radical, [XY] is the concentration of reactant X Y, [R] is the concentration of the 5-hexenyl radical, and [C] and [A] are the concentrations of cyclic and acylic products found at the end of the reaction. The concentration of 5-hexenyl radical in Eq. 1 cancels, and rearrangement gives Eq. 2. Note that Eq. 2 pertains to the situation where the concentration of X Y was great enough such that it effectively does not vary during the reaction (i.e.,
128 RADICALS
pseudo-first-order conditions). Note also that one needs to determine from independent studies that the products actually were formed in the radical reactions of interest and not by sequences that involved side reactions.
½A&=½C& ¼ ðkXY½XY&½R&Þ=ðkC½R&Þ |
ð1Þ |
kXY ¼ kCð½A&=½C&Þ½XY& 1 |
ð2Þ |
2.3. Electron Spin Resonance Spectroscopy
Electron spin resonance spectroscopy, also known as electron paramagnetic resonance or EPR spectroscopy, permits direct detection of radicals and has been employed extensively to determine structural features of radicals. Electron spin resonance is similar to nuclear magnetic resonance (NMR) spectroscopy, with which most chemists are quite familiar. In ESR, the spin of the electron is the counterpart to the odd nuclear spin in NMR. When the sample is in a magnetic field, the spin states of the odd electron differ in energy, and one measures energy absorbed by the sample when the low-energy-spin electrons are promoted to the higher energy level. The energy gaps in ESR are much larger than those in NMR, and high sensitivity is possible without the use of superconducting magnets. In relatively small magnetic fields of 3000–12,000 G, electrons absorb in the microwave region, 9000–34,000 MHz.
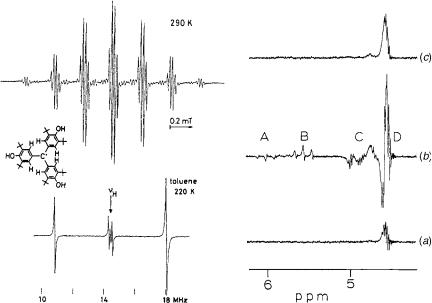
Much of the nomenclature of ESR spectroscopy is analogous to that in NMR spectroscopy. The g factor in ESR is similar to the chemical shift in NMR. Couplings in ESR spectroscopy refer to electron– electron couplings in polyradicals and are not important for most organic radicals. Hyperfine couplings in ESR are couplings of the electron with nuclear spins on atoms, and have direct analogy to nuclear spin couplings in NMR spectroscopy. For example, the ESR spectrum of the methyl radical appears as a 1:3:3:1 quartet due to hyperfine coupling of the electron with the three equivalent protons. The ESR spectrum25 of a triarylmethyl radical in Figure 4.4 demonstrates both the high resolution and hyperfine couplings. It is a well-resolved septet (6 ortho protons) of quartets (3 hydroxy protons).
ESR hyperfine couplings are listed as a values in analogy to the J values of NMR couplings. They are usually given in gauss (G), but they also can be given in millitesla (mT) or MHz (1 G ¼ 0.1 mT ¼ 2.80 MHz, when g ¼ 2:0023). They are quite large with the result that couplings of remote nuclei can be observed giving rich structural information about the radical. Absolute values of the hyperfine couplings in the ethyl radical, for example, are a ¼ 22:4 G for the a-protons and a ¼ 26:9 G for the b-protons where instrumental resolution is in the tenths of gauss, and hyperfine couplings to the g-protons in the cyclohexyl radical of a ¼ 0:5 and 0.7 G are readily observed.
2.3.1. Structural Information from ESR Spectroscopy. ESR spectroscopy provides considerable information about the structures of radicals. The identity of
IDENTIFICATION AND CHARACTERIZATION OF RADICALS |
129 |
Figure 4.4. Spectra of radicals. Left: ESR (top) and ENDOR (bottom) spectra of a triarylmethyl radical. [Reprinted with permission from B. Kirste, W. Harrer, and H. Kurreck, J. Am. Chem. Soc. 1985, 107, 20–28. Copyright # 1985 American Chemical Society.] Right: CIDNP effects in the vinyl proton absorbance region in a 60 MHz NMR spectrum from reaction of BuLi and BuBr before mixing (a), 30 s after mixing (b) and after completion of the reaction (c). [Reprinted with permission from H. R. Ward, and R. G. Lawler, J. Am. Chem. Soc. 1967, 89, 5518–5519. Copyright # 1967 American Chemical Society.]
the atom with the odd electron can be obtained from the g factor. For organic radicals, the g factor is close to that of a free electron. Structural information is obtained from hyperfine coupling constants.26
The hyperfine coupling between a nuclear spin and the electron spin at a radical center is a function of the amount of odd electron spin at the nucleus, which in turn is a function of the degree of s character in the orbital containing the odd electron. In the hydrogen atom, the electron is in the 1s orbital resulting in a maximum hyperfine interaction, and a ¼ 507 G. For the planar methyl radical, with the electron nominally residing in a 2p orbital, one might expect no 13C hyperfine coupling because the p orbital has a node at the nucleus. However, a coupling of a ¼ 39 G is observed for 13C in methyl due to orbital mixing that gives some s character to the orbital containing the odd electron. The maximum coupling possible for 13C is 1100 G, and the observed 39 g indicates somewhat <4% s character in the hybrid orbital. The formalism used is a ¼ A0(Cns), where A0 is the maximum coupling and Cns is the coefficient of the s orbital in the hybrid orbital.
Proton hyperfine couplings in the ESR spectrum of the methyl radical are 23 G, where the sign of the coupling is not important except for CIDNP effects. The
130 RADICALS
α-Coupling |
β-Coupling |
Figure 4.5. Origin of a- and b-proton hyperfine couplings.
electron spin density residing at the proton nuclei arises from spin polarization of the electrons in the CH bonds (Fig. 4.5). The electron in the CH bond that has spin parallel to that of the unpaired electron has increased electron density adjacent to the proton. The a value for the a protons on the carbon containing the odd electron in a p orbital are given by the McConnell equation (Eq. 3), where QHCH is a constant approximately equal to 28 G, and rC is the spin density on the carbon atom.
In the methyl radical, rC is unity, and jaðobsÞj ¼ 23 G. In the benzene radical anion, spin density is equally distributed on the six carbon atoms, rC is 0.17, and
jaðobsÞj ¼ 3:1 G for each proton.
aaH ¼ QCHH rC |
ð3Þ |
The protons on atoms adjacent to a radical center also couple with the unpaired electron, giving rise to b-proton hyperfine couplings (Fig. 4.5). These interactions are from orbital mixings that are equivalent to hyperconjugation of the CH bonding electrons with the radical center. The extent of mixing is a function of the dihedral angle (y) defined by the orbital of the radical, the atom of the radical center, the adjacent atom, and the hydrogen atom. A maximum in the b-proton hyperfine coupling in an alkyl radical will arise when the CH bond is parallel to the p orbital, and the coupling will be zero when the CH bond is perpendicular to the orbital.
Various qualitative equations have been used for b-proton hyperfine interactions,
but they all include a cos2 y term. In Eq. 4, the value of QHCHH is 50 G, and rC is again the spin density on the carbon atom. The Heller–McConnell equation (Eq. 5)
contains two constants; A is 5 G, and B is 40–50 G. The ESR spectrum is time averaged on the time scale of most conformational changes in acyclic radicals, and the value of aHb is a weighted average of the conformational populations. For a freely rotating methyl group in the position adjacent to the radical center, as in the ethyl radical, the integral of cos2y from 0 to 2p is 0.5, and the value of aHb predicted from Eq. 4 is 25 G; the observed value is aHb ¼ 27 G.
abH ¼ QCCHH rCðcos2 yÞ |
ð4Þ |
abH ¼ ðA þ B cos2 yÞrC |
ð5Þ |
g-Proton hyperfine couplings also have a cos2 y function, where the dihedral angle is defined by the orbital containing the odd electron and the CH bond. In the 1-adamantyl radical, the g protons with 180 dihedral angles and cos2 y ¼ 1
IDENTIFICATION AND CHARACTERIZATION OF RADICALS |
131 |
have a ¼ 3:08, and the g protons with 60 dihedral angles and cos2 y ¼ 0:25 have a ¼ 0:80 G.
The utility of ESR spectra for determining the structures of radicals is demonstrated by considering some examples. Methyl group substitution for hydrogen in the methyl radical ultimately results in slight deviation from planarity with a low inversion barrier. The 13C a values for methyl, ethyl, isopropyl, and tert-butyl are 38.3, 39.1, 41.3, and 45.2 G, respectively. The tert-butyl radical is indicated to have10 deviation from planarity, which is confirmed by infrared (IR) and Raman spectroscopy.1
Substituting fluorine atoms for hydrogen in the methyl radical results in increasing pyramidalization with increasing numbers of fluorine atoms as discussed in Section 1.1. For the series of radicals CH3, CH2F, CHF2, and CF3, the 13C hyperfine couplings are 38, 55, 146, and 272 G, respectively.27 Using the value of 1100 G for the coupling of 13C to an electron in a 2s orbital, one concludes that the odd electron in the trifluoromethyl radical resides in an orbital that has 25% s character (i.e., the radical is tetrahedral).3
For the vinyl radical, the 13C hyperfine coupling for the a-carbon is 107.6 G, which would suggest 10% s character in the hybrid orbital. The vinyl radical clearly has the odd electron in a s-type orbital because the b protons of the vinyl radical have distinct hyperfine couplings with a ¼ 37 G for the cis-H, and a ¼ 65 G for the trans-H. The cyclopropyl radical also is a s radical with an a-13C a value of
98 G, whereas the cyclohexyl radical, which has a nearly planar radical center, has an a-13C a value of 41 G.
2.4. Electron Nuclear Double Resonance Spectroscopy
Electron Nuclear Double Resonance (ENDOR) spectroscopy is a technique similar to double resonance NMR spectroscopic methods. In an ENDOR experiment, the sample is irradiated with a constant microwave frequency at the resonance energy of the unpaired electron. The intensity of the microwave beam is adjusted such that the ESR signal is partially saturated. The sample is then irradiated with radio frequency in the region of the NMR absorbances of the coupled nuclei in the magnetic field employed. As the radio frequency (rf) is varied, the energy absorbed in the microwave region is monitored. When the rf matches the resonance condition of a nucleus coupled to the unpaired electron, an Overhauser relaxation of the partially saturated electron spin occurs, and an increase in absorbance of microwave energy is observed. One obtains an output of ESR signal intensity as a function of rf (Fig. 4.4).
The hyperfine interactions of proton nuclei with the unpaired electron in a radical splits the proton signals into doublets with large coupling constants (up to 100 MHz) in comparison to the J values of 10 Hz observed in NMR spectroscopy. For the small magnetic field employed in an ESR spectrometer, all uncoupled protons absorb at effectively the same rf. The ENDOR spectrum appears as a series of doublets with lines spaced equally upfield and downfield from the expected absorbance of an uncoupled proton.